These alterations ultimately result in cell cycle arrest and apoptosis in melanoma cells. The HSP90 inhibitor ganetespib, with IC50 values,100 nM, exhibited profound antiproliferative activity against a panel of cutaneous melanoma cells including those that carry B-RAF and N-RAS mutations, as well as those with acquired resistance to BRAF inhibition. Ganetespib exerted its antiproliferative activity through induction of cell cycle arrest and apoptosis in association with inhibition of multiple tyrosine receptor kinases, B-RAF, CRAF, and the AKT and Erk1/2 pathways. The PI3K/Akt and MAPK/Erk pathways are critical for melanoma cell growth and survivaland were significantly inhibited by ganetespib. During the course of this study, similar effects of the HSP90 inhibitor XL888 on phosphorylation of Akt and Erk1/2 in melanoma cells have been reported. Inhibition of these pathways may contribute to ganetespib induced growth inhibition and apoptosis as inhibition of these pathways, alone or in combination, BKM120 reduced viability of K008 and K028 cellsand induced apoptosis in melanoma cells. In agreement with previous findings that Akt but not Erk1/2 was a client protein of HSP90, the expression of Akt but not Erk1/2 was reduced by ganetespib. Ganetespib may inhibit Akt phosphorylation through downregulating Akt expression and repress Erk1/2 phosphorylation through downregulating their upstream activating kinases, as MEK and 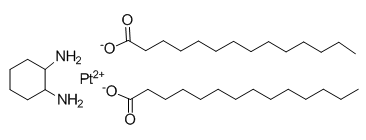 MEKK1 are client proteins of HSP90. Akt and Erk1/2 can be activated by BYL719 signals from multiple tyrosine kinase receptors including EGFR, IGF-1R and c-Met. In agreement with being client proteins of HSP90, the expression of these receptors was decreased by ganetespib. Downregulation of these receptors will also result in inhibition of Akt and Erk1/2 phosphorylation. Thus, ganetespib may inhibit Akt and Erk1/2 activation by targeting multiple cellular signaling processes. Small molecule inhibitors of c-Met, EGFR or IGF-1R reduced viability of K008 and K028 cells that express all these receptors, suggesting that these receptor tyrosine kinases may play a role in survival and growth of these cell lines and their inhibition may be relevant to the anti-melanoma activity of ganetespib. Ganetespib induced cell cycle arrest at G1and/or G2/M phase in cell line dependent manner. Similar cell cycle effects were also observed with XL888. Ganetespib induced cell cycle arrest was associated with upregulation of negative cell cycle regulatorsand/or downregulation of positive regulators. This is in general in line with the roles of these regulators in cell cycle regulation. CDK1, CDK2 and CDK4 have been reported to be chaperoned by HSP90. However, cyclin D1 and cyclin E are not considered to be a client protein for HSP90. The observed downregulation of cyclin D1 may result from downregulation of Akt and Erk1/2 pathways, which control cyclin D1 expression. Downregulation of cyclin E could result from decreased D-type cyclins, as the transcriptional activation of the cyclin E gene depends on the activity of D-type cyclins. The upregulation of the CDK inhibitors p27Kip1 and p21Cip1 could be attributed to inhibition of the Akt and Erk pathways. Despite being reported to be a client protein of HSP90and downregulated in K029 and K033 cells, cyclin B1 was induced by ganetespib in two cell linesthat were arrested at G2/M. Cyclin B1 activity is essential for progression from G2 into M phase. Binding of cyclin B1 to CDK1 allows CDK1 to be activated. The active cyclin B1-CDK1 complex translocates to the nucleus and phosphorylates nuclear substrates. These phosphorylation events are necessary for mitotic onset.
MEKK1 are client proteins of HSP90. Akt and Erk1/2 can be activated by BYL719 signals from multiple tyrosine kinase receptors including EGFR, IGF-1R and c-Met. In agreement with being client proteins of HSP90, the expression of these receptors was decreased by ganetespib. Downregulation of these receptors will also result in inhibition of Akt and Erk1/2 phosphorylation. Thus, ganetespib may inhibit Akt and Erk1/2 activation by targeting multiple cellular signaling processes. Small molecule inhibitors of c-Met, EGFR or IGF-1R reduced viability of K008 and K028 cells that express all these receptors, suggesting that these receptor tyrosine kinases may play a role in survival and growth of these cell lines and their inhibition may be relevant to the anti-melanoma activity of ganetespib. Ganetespib induced cell cycle arrest at G1and/or G2/M phase in cell line dependent manner. Similar cell cycle effects were also observed with XL888. Ganetespib induced cell cycle arrest was associated with upregulation of negative cell cycle regulatorsand/or downregulation of positive regulators. This is in general in line with the roles of these regulators in cell cycle regulation. CDK1, CDK2 and CDK4 have been reported to be chaperoned by HSP90. However, cyclin D1 and cyclin E are not considered to be a client protein for HSP90. The observed downregulation of cyclin D1 may result from downregulation of Akt and Erk1/2 pathways, which control cyclin D1 expression. Downregulation of cyclin E could result from decreased D-type cyclins, as the transcriptional activation of the cyclin E gene depends on the activity of D-type cyclins. The upregulation of the CDK inhibitors p27Kip1 and p21Cip1 could be attributed to inhibition of the Akt and Erk pathways. Despite being reported to be a client protein of HSP90and downregulated in K029 and K033 cells, cyclin B1 was induced by ganetespib in two cell linesthat were arrested at G2/M. Cyclin B1 activity is essential for progression from G2 into M phase. Binding of cyclin B1 to CDK1 allows CDK1 to be activated. The active cyclin B1-CDK1 complex translocates to the nucleus and phosphorylates nuclear substrates. These phosphorylation events are necessary for mitotic onset.
The most advanced molecule crizotinib which targets the receptor tyrosine kinases ALK and c-MET
In future studies, we propose to utilize the ferret model of IAV infection to assess the therapeutic potential of SAP given that ferrets express abundant a-linked SA throughout the respiratory tractand human IAV strains infect ferrets without the need for prior adaptation. Such studies would provide important insight regarding the ability of SAP to ameliorate influenza disease severity and/ or virus transmission in a model that more accurately depicts many characteristics of human disease. Members of the transforming growth factor-betasuperfamily bind transmembrane receptor serine/threonine kinases to activate Smad and non-Smad pathways for the control of normal development and tissue repair. Ligand binding induces type II receptor phosphorylation of associated type I receptors, leading to Smad recruitment and phosphorylation by the type I receptor. The receptor-associated Smadssubsequently SCH727965 779353-01-4 assemble with co-Smad4 for nuclear transport and transcriptional activation. Small molecule inhibitors of the type I receptorshave proved to be valuable pharmacological tools to characterize TGF-b and BMP pathways in signaling, as well as stem cell biology. TGF-b inhibitors such as SB-431542 inhibit Smad2/3 phosphorylation by ALK4, ALK5 and ALK7, as well as non-classical Smad1/5 phosphorylation by ALK5. Conversely, inhibitors of BMP signaling have recently been described that specifically inhibit Smad1/5/8 phosphorylation by ALK1, ALK2, ALK3 and ALK6. Notably, these molecules have shown efficacy in a variety of disease models, including chronic anemia, prostate cancer, muscle wasting, heterotopic ossification, atherosclerosis and vascular calcification. While specific TGF-b inhibitors have been developed over many years, BMP inhibitor development remains at an early stage. New leads in this target area are desirable for several reasons. First, current work follows a single high throughput screen performed in the zebrafish system. Second, independent tool compounds are preferred for functional validation, whereas the screening hit dorsomorphin, and derivatives DMH1and LDN-193189, share the same pyrazolopyrimidine scaffold. Third, more selective compounds are needed to minimize unwanted off-target effects. Most importantly, there is an urgent need for selective ALK2 inhibitors to treat the debilitating bone disorder fibrodysplasia ossificans progressiva. FOP sufferers carry a gain of function mutation in the intracellular domain of ALK2, resulting in episodic bone formation in skeletal muscle and connective tissue that ultimately renders movement impossible. U0126 molecular weight Trauma and surgery only accelerate the condition, while biological inhibitors lacking cell penetrance are ineffective. As an alternative but 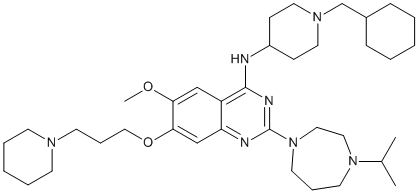 complementary strategy to phenotypic screens, we used direct screening of recombinant human kinases to identify new inhibitor leads against ALK2. We report a novel BMP inhibitor scaffold, comprising a 2-aminopyridine core and a trimethoxyphenyl specificity group, which is both potent and selective. The identified inhibitor K02288 provides a new pharmacological tool to investigate the diversity of BMP signaling in both normal and pathobiology. The development of selective small molecule inhibitors of protein kinases presents a major challenge due to the high sequence conservation of the ATP pocket. Here we report a novel 2-aminopyridine inhibitor K02288 with potent and selective activity against type I BMP receptor kinases. The 2-aminopyridine group is an ATP-mimetic that binds the kinase hinge region through two conserved hydrogen bonds. Importantly, selective kinase inhibitors with this scaffold have been identified previously.
complementary strategy to phenotypic screens, we used direct screening of recombinant human kinases to identify new inhibitor leads against ALK2. We report a novel BMP inhibitor scaffold, comprising a 2-aminopyridine core and a trimethoxyphenyl specificity group, which is both potent and selective. The identified inhibitor K02288 provides a new pharmacological tool to investigate the diversity of BMP signaling in both normal and pathobiology. The development of selective small molecule inhibitors of protein kinases presents a major challenge due to the high sequence conservation of the ATP pocket. Here we report a novel 2-aminopyridine inhibitor K02288 with potent and selective activity against type I BMP receptor kinases. The 2-aminopyridine group is an ATP-mimetic that binds the kinase hinge region through two conserved hydrogen bonds. Importantly, selective kinase inhibitors with this scaffold have been identified previously.
Indicating that the partial decrease was not due to cleavage at basic residuesare elevated by bortezomib treatment
In addition, many of the BMS-907351 peptides that are generated by cleavage at hydrophobic residuesare also elevated by the bortezomib treatment. This finding is much different than the results with epoxomicin, a proteasome inhibitor that also shows greatest potency towards the beta-5 site, and which generally causes a decrease in levels of peptides that require cleavage at beta5 sites. Because our previous data with epoxomicin did not include all of the peptides found in the present study, we reanalyzed the epoxomicin data to search for every peptide found in the present study; those that were found are included together with the previously published epoxomicin data in the heat maps in Figure 3. Although both bortezomib and epoxomicin are potent inhibitors of the beta5 proteasome subunit, there are dozens of likely beta-5 products that are decreased by epoxomicin treatment but elevated by bortezomib treatment. Sorting the heat map data by protein precursor reveals that many of the peptides elevated by bortezomib treatment arise from just six proteins: 60S acidic ribosomal protein P2, heat shock 10kDa protein 1, histidine triade nucleotide-binding protein 1, macrophage migration inhibitory factor, nucleophosmin, and protein SET. Collectively, these six proteins account for 65 of the peptides that are greatly elevated upon bortezomib treatment. Previously, it was noted that many of the observed cellular peptides were derived from cytosolic proteins, although peptides were also found that corresponded to mitochondrial and nuclear proteins. To test if the cellular location of the protein correlated with changes in peptides, the number of peptides in each group were compared. As previously found, most of the peptides detected in the present study were derived from cytosolic proteins. Over 100 of these cytosolic protein-derived peptides were greatly elevated by bortezomib treatment, but this represented only,30% of the total number of cytosolic protein-derived peptides, and many peptides derived from cytosolic proteins showed no change or decreased upon treatment. In contrast, the majority of the mitochondrial protein-derived peptides showed a very large increase upon bortezomib treatment, and only a handful did not change or showed a decrease. In previous studies, approximately 50% of the cellular peptides in HEK293T and other  cell lines were found to represent the Nor C-terminus of the protein. In the present study, approximately 70% of peptides unaffected by bortezomib were Nor C-terminal peptides and only 30% represented internal peptides. In contrast,,80% of the peptides which were greatly elevated by bortezomib represented internal fragments of the proteins. For the analysis shown in Figure 5, both 50 and 500 nM treatments were combined. To determine if the peptides that showed a partial decrease or increase were comparable between these two concentrations of bortezomib, the two groups were analyzed separately. For this analysis, only those peptides detected in both the 50 and 500 nM treatment groups were considered. The 26 peptides which showed a partial Ibrutinib decreasein the 500 nM bortezomib group were similarly affected by treatment with 50 nM bortezomib, indicating that the partial effect was not due to incomplete inhibition of bortezomib at the lower dose. However, those peptides partially increased by 50 nM bortezomib showed a significantly larger increase upon treatment with 500 nM drug. Similar analysis was performed to compare peptides that partially decreased or increased upon treatment with 500 nM bortezomib for 30 or 90 minutes. Levels of peptides that partially decreased after 30 minutes of treatment were not significantly different after 90 minutes of treatment.
cell lines were found to represent the Nor C-terminus of the protein. In the present study, approximately 70% of peptides unaffected by bortezomib were Nor C-terminal peptides and only 30% represented internal peptides. In contrast,,80% of the peptides which were greatly elevated by bortezomib represented internal fragments of the proteins. For the analysis shown in Figure 5, both 50 and 500 nM treatments were combined. To determine if the peptides that showed a partial decrease or increase were comparable between these two concentrations of bortezomib, the two groups were analyzed separately. For this analysis, only those peptides detected in both the 50 and 500 nM treatment groups were considered. The 26 peptides which showed a partial Ibrutinib decreasein the 500 nM bortezomib group were similarly affected by treatment with 50 nM bortezomib, indicating that the partial effect was not due to incomplete inhibition of bortezomib at the lower dose. However, those peptides partially increased by 50 nM bortezomib showed a significantly larger increase upon treatment with 500 nM drug. Similar analysis was performed to compare peptides that partially decreased or increased upon treatment with 500 nM bortezomib for 30 or 90 minutes. Levels of peptides that partially decreased after 30 minutes of treatment were not significantly different after 90 minutes of treatment.
Therefore there are two possible mechanisms by which these inhibitors may prevent cleavage of substrate
The resulting active enzyme is a dimer, wherein each subunit contains a p10 and p20 chain and one active site. The caspase enzymatic mechanism is similar to other cysteine proteases; substrate binds to the active site to form the Michaelis complex, a covalent tetrahedral intermediate is formed by attack of the active-site thiolate cysteine on the scissile carbonyl, the substrate amide bond is cleaved to generate an acyl enzyme intermediate, and the intermediate is hydrolyzed by water to yield the new substrate C-terminus and apo-enzyme. Active caspases are capable of cleaving numerous cellular proteinsand carrying out the terminal phase of cell death signaling. Due to the role of caspase-6 in neurodegeneration, there is strong interest in developing selective, small-molecule inhibitors of this enzyme. This family of proteases has proven resistant to traditional methods of drug discovery, however, and most known inhibitors contain a covalent warhead, significant peptidic character, and/or an aspartic acid. Each of these characteristics reduces the potential for caspase selectivity, cell permeability, and blood-brain barrier penetrance. For instance, the traditional caspase probes used in biological assays are tetrapeptides containing the ideal substrate sequences for each caspase and a covalent warhead that reversibly or irreversibly modifies the active-site cysteine. These tools lack the necessary caspase selectivity profiles to facilitate the delineation of isoformspecific signaling pathways in a cellular context. To address these challenges, a number of alternative chemical approaches have been used. Leyva, et al, recently disclosed the design of novel, BMS-907351 nonpeptidic inhibitors identified through “substrate assisted screening”; while potent, these compounds are non-selective and still contain an irreversible covalent warhead. There has also been significant interest in developing noncompetitive or allosteric inhibitors, with the idea that non-active site binding could achieve greater selectivity and improved physicochemical properties over competitive inhibitors. This notion is supported by the discovery of an allosteric site at the dimer interface of caspases 1, 3, and 7. Applying the disulfide-trappingmethod of fragment discovery, scientists at Sunesis Pharmaceuticals identified fragments that bound at the dimer interface and inhibited enzymatic activity. These fragments were not tested for cellular activity, and the druggability of this site remains an interesting, open question. Using a fluorogenic assay platform we identified a series of molecules that inhibit caspase-6 in an unexpected and mechanistically uncompetitive fashion. Detailed structural and mechanistic studies with the most potent of these compounds  indicate that it binds to the enzyme-substrate complex in a highly specific manner to inhibit substrate turnover. This uncompetitive mechanism of enzyme inhibition is novel for any of the caspase family members. The present compound demonstrates a very distinctive molecular recognition for caspase-6/VEID peptides, and points the way towards utilizing uncompetitive inhibition as a strategy for the discovery of highly selective caspase inhibitors. Our search for caspase-6 inhibitors led to the identification of a highly selective molecule that Compound Library inhibits the enzyme via a novel mechanism not previously described for any of the caspases. Although it has recently been demonstrated for another cysteine protease that the acyl-enzyme intermediate is the primary resting state during the catalytic cycle, stabilization of this intermediate by 3 can be ruled out as the sole mechanism of inhibition, since no fluorophore dependence would be expected if this were the case.
indicate that it binds to the enzyme-substrate complex in a highly specific manner to inhibit substrate turnover. This uncompetitive mechanism of enzyme inhibition is novel for any of the caspase family members. The present compound demonstrates a very distinctive molecular recognition for caspase-6/VEID peptides, and points the way towards utilizing uncompetitive inhibition as a strategy for the discovery of highly selective caspase inhibitors. Our search for caspase-6 inhibitors led to the identification of a highly selective molecule that Compound Library inhibits the enzyme via a novel mechanism not previously described for any of the caspases. Although it has recently been demonstrated for another cysteine protease that the acyl-enzyme intermediate is the primary resting state during the catalytic cycle, stabilization of this intermediate by 3 can be ruled out as the sole mechanism of inhibition, since no fluorophore dependence would be expected if this were the case.
To improve outcomes for patients with certain cancer types either as combined with chemotherapy
However, only a fraction of treated patients typically derive clinical benefit. Predictive biomarkers identifying patients most likely to respond would allow for a more targeted approach to treatment and, therefore, would be of significant clinical value. To date, no validated BU 4061T biomarker has been identified for any angiogenesis inhibitor despite extensive investigation. Lambrechts et al recently described efforts to identify predictive biomarkers for bevacizumab: although potential markers have been identified in certain tumor types, as yet none have proven robust. A recent prospective study found an association between low VEGF-A levels and both progression-free survival and overall survival in patients with nonsquamous NSCLC. However, because the study did not include a control arm it was not possible to differentiate between prognostic and predictive value of the biomarker. Motesanib is a potent small-molecule inhibitor of VEGF receptors 1, 2, and 3; platelet-derived growth factor receptor; and Kit, with demonstrated antitumor activity as monotherapy and in combination with chemotherapy. In the motesanib first-in-human study, analysis of potential biomarker candidates showed a strong pharmacodynamic response of placental growth factor and further suggested that increased levels of PLGF from baseline were associated with increased motesanib exposure and possibly correlated with tumor shrinkage. PLGF is a VEGF-A homolog and a VEGFR1 ligand that is up-regulated during hypoxia, and may be involved in pathologic angiogenesis, possibly by increasing the responsiveness of endothelial cells to VEGF-A. The increase in PLGF following motesanib treatment possibly represents a compensatory upregulation in response to VEGF pathway blockade. Subsequent phase 2 studies with motesanib showed a consistent association between increased levels from baseline in PLGF and outcomes across different tumor types, including thyroid cancer, breast cancer, and non�Csmall-cell lung cancer. LDK378 Furthermore, other inhibitors of the VEGF pathway have been known to induce pharmacodynamic changes in PLGF, which, in some cases, have been associated with outcomes including objective response and OS. Taken together, the data suggested that PLGF may serve as a biomarker for the biologic effect of VEGF receptor inhibitors, and as such, it may be a potential biomarker identifying a population most likely to benefit from continued treatment with these agents. The PLGF data collected in motesanib phase 2 studies formed a strong body of evidence that supported further prospective testing of PLGF as a potential biomarker in the large international phase 3 Motesanib NSCLC Efficacy and Tolerability study of motesanib plus carboplatin/paclitaxel versus placebo plus carboplatin/paclitaxel in patients with nonsquamous NSCLC. However, the study did not  meet its primary endpoint, and PLGF analysis with a validated assay developed specifically as a companion diagnostic test did not reveal an association between change from baseline in PLGF and OS. To date, MONET1 remains the only large, prospective study of a biomarker candidate for an angiogenesis inhibitor. Considering the body of evidence for PLGF as a biomarker for motesanib and the rigorous analysis of data that formed the basis of the PLGF hypothesis for MONET1, the study’s negative biomarker results demonstrate the challenges in the development of a valid predictive biomarker. Here we describe the processes we undertook in an effort to develop PLGF as a pharmacodynamic biomarker for motesanib using an ongoing phase 3 study of motesanib in patients with NSCLC and supporting data from the preceding phase 2 study of motesanib in NSCLC.
meet its primary endpoint, and PLGF analysis with a validated assay developed specifically as a companion diagnostic test did not reveal an association between change from baseline in PLGF and OS. To date, MONET1 remains the only large, prospective study of a biomarker candidate for an angiogenesis inhibitor. Considering the body of evidence for PLGF as a biomarker for motesanib and the rigorous analysis of data that formed the basis of the PLGF hypothesis for MONET1, the study’s negative biomarker results demonstrate the challenges in the development of a valid predictive biomarker. Here we describe the processes we undertook in an effort to develop PLGF as a pharmacodynamic biomarker for motesanib using an ongoing phase 3 study of motesanib in patients with NSCLC and supporting data from the preceding phase 2 study of motesanib in NSCLC.