Recently, in contrast to our speculation, Chauhan et al. reported the effectiveness of MLN9708 to overcome bortezomib resistance. As several mechanisms have been proposed for bortezomib resistance in addition to ?5 subunit mutations, MLN9708 may be effective for such cases. HPDs are expected to compensate for the weak points of bortezomib as well as the second generation PIs described above, because HPDs are non-peptide agents that inhibit all three catalytic subunits of the proteasome with equal kinetics and could be orally bioactive. Moreover, crystal structure analyses indicate that the binding mode is completely different from that of bortezomib and NPI-0052. This ensures the activity of this agent against Nutlin-3 bortezomib-resistant cells, which was experimentally proven in this study, and probably against cells developing the resistance to NPI-0052. Moreover, we have found that oral administration of K-7174 is indeed effective and is not associated with obvious toxicities, including leukocytopenia, in a murine xenograft model. These features provide a rationale 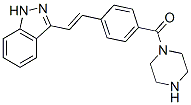 for the clinical translation of HPDs as novel PIs with effectiveness for the Rapamycin mTOR inhibitor treatment of bortezomibresistant patients, a low probability of acquired drug resistance, and flexibility in dosing schedules. In a plethora of in vitro studies it has been extensively demonstrated that inhibition of the proteasome for instance by the tripeptide aldehyd MG-132 or the dipeptide boronate bortezomib selectively kills tumor cells of varying origin. Proteasomal inhibitors also sensitize cells to radio- and chemotherapy and even to apoptosis induced by death receptor ligands. However, as the proteasome targets not only pro-, but also anti-apoptotic proteins, a successful combination therapy requires a successive application of first the apoptosis-inducing agent ensuring the breakdown of anti-apoptotic proteins followed by the PI treatment that then prevents degradation of the generated pro-apoptotic proteins. Nevertheless, bortezomib was the first PI used in clinical trials and approved to treat patients suffering from multiple myeloma or mantle cell lymphoma. Although the new generation of proteasome inhibitors such as salinosporamide and carfilzomib appear to exhibit somewhat different mechanisms of action than bortezomib, central to apoptosis induction by many PIs is certainly the mitochondrial or intrinsic death pathway, as their cytotoxic activity is almost completely abrogated in cells deficient for Bax and Bak. Consistently, a number of studies strongly implicated certain proapoptotic BH3-only proteins in PI-induced apoptosis. For instance, the pro-apoptotic cleavage product of Bid, t-Bid, is degraded by the proteasome and treatment of HeLa cells with MG-132 resulted in accumulation of t-Bid and sensitized the cells to death receptor-induced apoptosis. Also Bik and Bim were found to be upregulated following PI treatment and cells deficient for both or cells in which Bik and Bim were down regulated by RNA interference were refractory to its cytotoxic action. Likewise, different PIs including bortezomib and MG-132 were shown to induce expression of Noxa in several tumor models both at the protein and mRNA level and siRNA-mediated knockdown of Noxa partially rescued various tumor cells from PI-induced apoptosis. Expression of other Bcl-2 family members such as Puma, Bax, Bak, Bcl-2, and Bcl-XL remained mostly unaffected following treatment of different cell lines with PIs.
for the clinical translation of HPDs as novel PIs with effectiveness for the Rapamycin mTOR inhibitor treatment of bortezomibresistant patients, a low probability of acquired drug resistance, and flexibility in dosing schedules. In a plethora of in vitro studies it has been extensively demonstrated that inhibition of the proteasome for instance by the tripeptide aldehyd MG-132 or the dipeptide boronate bortezomib selectively kills tumor cells of varying origin. Proteasomal inhibitors also sensitize cells to radio- and chemotherapy and even to apoptosis induced by death receptor ligands. However, as the proteasome targets not only pro-, but also anti-apoptotic proteins, a successful combination therapy requires a successive application of first the apoptosis-inducing agent ensuring the breakdown of anti-apoptotic proteins followed by the PI treatment that then prevents degradation of the generated pro-apoptotic proteins. Nevertheless, bortezomib was the first PI used in clinical trials and approved to treat patients suffering from multiple myeloma or mantle cell lymphoma. Although the new generation of proteasome inhibitors such as salinosporamide and carfilzomib appear to exhibit somewhat different mechanisms of action than bortezomib, central to apoptosis induction by many PIs is certainly the mitochondrial or intrinsic death pathway, as their cytotoxic activity is almost completely abrogated in cells deficient for Bax and Bak. Consistently, a number of studies strongly implicated certain proapoptotic BH3-only proteins in PI-induced apoptosis. For instance, the pro-apoptotic cleavage product of Bid, t-Bid, is degraded by the proteasome and treatment of HeLa cells with MG-132 resulted in accumulation of t-Bid and sensitized the cells to death receptor-induced apoptosis. Also Bik and Bim were found to be upregulated following PI treatment and cells deficient for both or cells in which Bik and Bim were down regulated by RNA interference were refractory to its cytotoxic action. Likewise, different PIs including bortezomib and MG-132 were shown to induce expression of Noxa in several tumor models both at the protein and mRNA level and siRNA-mediated knockdown of Noxa partially rescued various tumor cells from PI-induced apoptosis. Expression of other Bcl-2 family members such as Puma, Bax, Bak, Bcl-2, and Bcl-XL remained mostly unaffected following treatment of different cell lines with PIs.
The theoretical add suggesting the importance of maintaining physiological levels of circulating FGF23
Findings that in ZDF rats proteinuria preceded FGF23 upregulation in the kidney and that the antiproteinuric effect of ACE inhibitor was associated with reduced FGF23 expression, could be taken to suggest proteinuria as a potential trigger of renal FGF23. Future studies are needed to address this possibility. In this context, a recent study demonstrated that in patients with CKD FGF23 was positively associated with proteinuria. Consistenly, we have documented here a positive correlation between renal FGF23 expression and proteinuria in diabetic rats. In conclusion, our data indicate that in experimental type 2 diabetes 1. the kidney is a site of FGF23 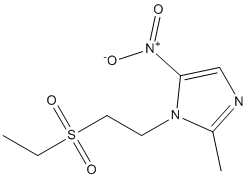 production; 2. during the progression of the disease, renal FGF23 increased in the face of Klotho and NaPi-2a co-transporter reduction; 3. ACE inhibitor therapy besides exhibiting antiproteinuric and renoprotective actions, attenuated FGF23 renal production and preserved the expression of Klotho resulting in sustained improvement of phosphate homeostasis. These data may offer new clues to understand how to interfere with the delicate balance of FGF23 and phosphorus in diabetes with potential implications in clinics. Glioblastoma multiforme is the most common and aggressive brain tumor in humans, and despite technical advances in neurosurgery and clinical neuro-oncology, the prognosis for GBM patients remains very poor. Most patients die within one year of diagnosis and are generally insensitive to current therapeutic genotoxic interventions. In the majority of GBM cases, resistance to such genotoxic modalities has been attributed to the attenuation of p53 function by alterations within the p53 signalling axis, including the overexpression of Murine Double Minute-2. The MDM2 oncoprotein, a major physiological negative regulator of p53, can bind to the p53 transactivation domain and interfere with the transcriptional regulatory mechanisms of p53. MDM2 is also an E3 ubiquitin ligase that promotes p53 proteasomal degradation. For this reason, inhibition of the interaction between MDM2 and p53 to reactivate endogenous p53 activity offers the opportunity for therapeutic intervention, particularly in GBMs. In GBMs, the p53 gene is relatively infrequently mutated; however, wild-type p53 remains dysfunctional due to overexpressed MDM2. Intensive work on different classes of MDM2 inhibitors has proven their therapeutic utility as activators of p53 in multiple tumor models. Indeed, it has been demonstrated that a number of small-molecule MDM2 inhibitors can disrupt the MDM2-p53 interaction, release p53 from negative control and activate the p53 pathway, leading to cell cycle arrest and apoptosis in a number of solid cancers and haematological malignancies. Moreover, many laboratories have shown that MDM2 inhibitors can synergise with conventional chemotherapeutic agents, resulting in enhanced efficacy. Interestingly, MDM2 inhibitors have been reported to induce cancer cell apoptosis even without the concomitant application of genotoxic stimuli. Little is known about the effects of MDM2 inhibitors on the in vitro growth of GBM cells. Recently, Nutlin-3, the first potent MDM2 small-molecule inhibitor identified, and new D-peptide derivatives were reported to be effective at inhibiting GBM cell growth in vitro, suggesting the validity of this experimental approach for the treatment of GBM. In the present study, we investigated the responsiveness of human GBM cell lines to a novel small-molecule MDM2 inhibitor with a spirooxoindolepyrrolidine core structure, named ISA27, which has been recently shown by nuclear magnetic resonance Everolimus mTOR inhibitor analysis to efficiently dissociate the reconstituted human MDM2-p53 complex. Consistently, ISA27 activated the p53 pathway in GBM cells and elicited the dose- and time-dependent inhibition of cell growth. ISA27 induced apoptosis and evoked cellular senescence, indicating that ISA27 promotes a pleiotropic anticancer effect in the GBM cells. The administration of ISA27 in vivo efficiently inhibited tumor growth in nude mice bearing a human GBM Bortezomib abmole bioscience xenograft. Significantly, ISA27 was non-toxic both in vitro in a normal human cell model and in vivo in a mouse model. A graphical assessment of synergy with regard to growth inhibition was performed using isobolographic analysis. In an isobologram, the equi-effective pairs of doses of two drugs are represented using rectangular coordinates. In the present study, the dose of ISA27 required to give a 50% effect was plotted on the abscissa, and the iso-effective dose of Temozolomide was plotted on the ordinate.
production; 2. during the progression of the disease, renal FGF23 increased in the face of Klotho and NaPi-2a co-transporter reduction; 3. ACE inhibitor therapy besides exhibiting antiproteinuric and renoprotective actions, attenuated FGF23 renal production and preserved the expression of Klotho resulting in sustained improvement of phosphate homeostasis. These data may offer new clues to understand how to interfere with the delicate balance of FGF23 and phosphorus in diabetes with potential implications in clinics. Glioblastoma multiforme is the most common and aggressive brain tumor in humans, and despite technical advances in neurosurgery and clinical neuro-oncology, the prognosis for GBM patients remains very poor. Most patients die within one year of diagnosis and are generally insensitive to current therapeutic genotoxic interventions. In the majority of GBM cases, resistance to such genotoxic modalities has been attributed to the attenuation of p53 function by alterations within the p53 signalling axis, including the overexpression of Murine Double Minute-2. The MDM2 oncoprotein, a major physiological negative regulator of p53, can bind to the p53 transactivation domain and interfere with the transcriptional regulatory mechanisms of p53. MDM2 is also an E3 ubiquitin ligase that promotes p53 proteasomal degradation. For this reason, inhibition of the interaction between MDM2 and p53 to reactivate endogenous p53 activity offers the opportunity for therapeutic intervention, particularly in GBMs. In GBMs, the p53 gene is relatively infrequently mutated; however, wild-type p53 remains dysfunctional due to overexpressed MDM2. Intensive work on different classes of MDM2 inhibitors has proven their therapeutic utility as activators of p53 in multiple tumor models. Indeed, it has been demonstrated that a number of small-molecule MDM2 inhibitors can disrupt the MDM2-p53 interaction, release p53 from negative control and activate the p53 pathway, leading to cell cycle arrest and apoptosis in a number of solid cancers and haematological malignancies. Moreover, many laboratories have shown that MDM2 inhibitors can synergise with conventional chemotherapeutic agents, resulting in enhanced efficacy. Interestingly, MDM2 inhibitors have been reported to induce cancer cell apoptosis even without the concomitant application of genotoxic stimuli. Little is known about the effects of MDM2 inhibitors on the in vitro growth of GBM cells. Recently, Nutlin-3, the first potent MDM2 small-molecule inhibitor identified, and new D-peptide derivatives were reported to be effective at inhibiting GBM cell growth in vitro, suggesting the validity of this experimental approach for the treatment of GBM. In the present study, we investigated the responsiveness of human GBM cell lines to a novel small-molecule MDM2 inhibitor with a spirooxoindolepyrrolidine core structure, named ISA27, which has been recently shown by nuclear magnetic resonance Everolimus mTOR inhibitor analysis to efficiently dissociate the reconstituted human MDM2-p53 complex. Consistently, ISA27 activated the p53 pathway in GBM cells and elicited the dose- and time-dependent inhibition of cell growth. ISA27 induced apoptosis and evoked cellular senescence, indicating that ISA27 promotes a pleiotropic anticancer effect in the GBM cells. The administration of ISA27 in vivo efficiently inhibited tumor growth in nude mice bearing a human GBM Bortezomib abmole bioscience xenograft. Significantly, ISA27 was non-toxic both in vitro in a normal human cell model and in vivo in a mouse model. A graphical assessment of synergy with regard to growth inhibition was performed using isobolographic analysis. In an isobologram, the equi-effective pairs of doses of two drugs are represented using rectangular coordinates. In the present study, the dose of ISA27 required to give a 50% effect was plotted on the abscissa, and the iso-effective dose of Temozolomide was plotted on the ordinate.
Treatment of RMS includes the use of intensive chemotherapeutic regimens in combination with surgical and radiation therapy
responsible for the prostaglandins essential for normal mucosal physiology in gut. As no gastrointestinal toxicity data were collected in this study, whether these phytochemicals cause gastrointestinal bleeding is still unknown and further study in these areas is required. Rhabdomyosarcoma is the most common soft tissue sarcoma in childhood, accounting for about 3% of all childhood tumors. This strategy has improved the CPI-613 survival rate for patients with localized disease to 70% albeit with significant toxicity. Despite aggressive multimodal therapy, high risk patients continue to have a poor prognosis with overall survival rates of 20�C30%. Therefore, there remains a great need for new therapies targeting the molecular pathways which are found to be altered in RMS. RMS tumors typically arise from skeletal muscle and are categorized as either of the alveolar or embryonal subtype based on their histology. ARMS tumors are driven by a translocation involving chromosome 2 or 1 with chromosome 13, resulting in the production of the fusion oncogene PAX3- or PAX7-FOXO1, respectively. In contrast, ERMS tumors commonly harbor loss of heterozygosity at 11p15.5 as well as point mutations in TP53, NRAS, KRAS, HRAS, PIK3CA and FGFR4 genes. Fibroblast Growth Factor Receptor 4, a FGF receptor family member, is a receptor tyrosine kinase that is implicated in the differentiation of myoblasts into skeletal muscle and muscle regeneration after injury. Highlighting a potential role in RMS, early microarray studies of RMS cell lines and tumors showed massive overexpression of FGFR4 and subsequent work showed that FGFR4 is a direct transcriptional target of the PAX3-FOXO1 fusion protein. Of note, recent sequencing studies identified activating mutations specific to FGFR4 in 7.5% of RMS tumors. These mutations occur at amino acid 535 and 550 of the kinase domain and promote tumor growth and metastasis in vivo by constitutively activating FGFR4. These reports emphasize the importance of FGFR4 in RMS and establish this cell surface tyrosine kinase receptor as a candidate target for RMS therapy. Ponatinib is an orally administered  tyrosine kinase inhibitor that was initially developed as an inhibitor for native and mutant forms of BCR-ABL. Recently, this therapy received accelerated FDA approval for the treatment of adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia and chronic phase, accelerated phase, or blast phase chronic myeloid leukemia who are resistant or intolerant to prior tyrosine kinase inhibitor therapy. The inhibition profile of ponatinib includes several other tyrosine Silmitasertib kinases, including FLT3, SRC, KIT, PDGFR, and FGFR. Of note, ponatinib has been shown to inhibit all four members of the FGFR family with an IC50 of less than 40 nM. Inhibition of FGFR family members by ponatinib has been demonstrated in preclinical models of endometrial cancers with FGFR2 mutations, bladder cancers with FGFR3 mutations, as well as breast, lung, and colon cancer cell lines harboring amplification of the FGFR1 or FGFR2 gene. In this study, a panel of RMS cell lines as well as a Ba/F3 cell line engineered to overexpress FGFR4 were tested for sensitivity to five FGFR tyrosine kinase inhibitors, including AP24534, AZD2171, BIBF1120, TKI258, and PHA739358. Of these, ponatinib was found to be the most potent FGFR4 inhibitor, inhibiting both wild-type and mutated FGFR4 phosphorylation and cell growth. Ponatinib also inhibited growth of tumors expressing mutated FGFR4 in vivo. Therefore, our results indicate that ponatinib is an effective FDA-approved drug which has the potential to treat RMS with overexpressed or mutated FGFR4. Alteration of FGFR4 signaling is a common mechanism of oncogenesis in both fusion positive and fusion negative rhabdomyosarcoma. Thus far, at least three mechanisms have been reported to result in the gain of function of FGFR4 in RMS. First, elevated FGFR4 expression in RMS tumors can be a direct result of the PAX3-FOXO1 fusion oncogene, since FGFR4 was reported to be one of the direct targets of the transcription factor. Secondly, up-regulation of FGFR4 expression in RMS can be achieved through localized gene amplification. Thirdly, 7.5% of primary RMS tumors harbor a damaging missense mutation in the tyrosine kinase domain of FGFR4 which results in a constitutively active signaling molecule. The first two mechanisms result in elevated expression of wild-type FGFR4 in RMS, which is both common and associated with poor outcome.
tyrosine kinase inhibitor that was initially developed as an inhibitor for native and mutant forms of BCR-ABL. Recently, this therapy received accelerated FDA approval for the treatment of adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia and chronic phase, accelerated phase, or blast phase chronic myeloid leukemia who are resistant or intolerant to prior tyrosine kinase inhibitor therapy. The inhibition profile of ponatinib includes several other tyrosine Silmitasertib kinases, including FLT3, SRC, KIT, PDGFR, and FGFR. Of note, ponatinib has been shown to inhibit all four members of the FGFR family with an IC50 of less than 40 nM. Inhibition of FGFR family members by ponatinib has been demonstrated in preclinical models of endometrial cancers with FGFR2 mutations, bladder cancers with FGFR3 mutations, as well as breast, lung, and colon cancer cell lines harboring amplification of the FGFR1 or FGFR2 gene. In this study, a panel of RMS cell lines as well as a Ba/F3 cell line engineered to overexpress FGFR4 were tested for sensitivity to five FGFR tyrosine kinase inhibitors, including AP24534, AZD2171, BIBF1120, TKI258, and PHA739358. Of these, ponatinib was found to be the most potent FGFR4 inhibitor, inhibiting both wild-type and mutated FGFR4 phosphorylation and cell growth. Ponatinib also inhibited growth of tumors expressing mutated FGFR4 in vivo. Therefore, our results indicate that ponatinib is an effective FDA-approved drug which has the potential to treat RMS with overexpressed or mutated FGFR4. Alteration of FGFR4 signaling is a common mechanism of oncogenesis in both fusion positive and fusion negative rhabdomyosarcoma. Thus far, at least three mechanisms have been reported to result in the gain of function of FGFR4 in RMS. First, elevated FGFR4 expression in RMS tumors can be a direct result of the PAX3-FOXO1 fusion oncogene, since FGFR4 was reported to be one of the direct targets of the transcription factor. Secondly, up-regulation of FGFR4 expression in RMS can be achieved through localized gene amplification. Thirdly, 7.5% of primary RMS tumors harbor a damaging missense mutation in the tyrosine kinase domain of FGFR4 which results in a constitutively active signaling molecule. The first two mechanisms result in elevated expression of wild-type FGFR4 in RMS, which is both common and associated with poor outcome.
inhibition in COS-7 and BEAS-2B cells might suggest that pitstop is acting in all cases through its effect in blocking
To examine whether VE-821 ATM/ATR inhibitor pitstop 2 is inhibiting CIE through its effects on the Axitinib clathrin N-terminal domain, we looked at transferrin and MHCI endocytosis in cells depleted of clathrin heavy chain or the m2 subunit of the adaptor protein complex AP2, both of which were depleted to approximately 12 and 14% of control levels, respectively. Depletion of the m2 subunit of AP2or of clathrin heavy chainby siRNA results in a block in transferrin endocytosis in most cells while endocytosis of MHCI by CIE is not affected. The addition of pitstop 2  to the m2 and clathrin heavy chain depleted cells still led to a block in endocytosis of MHCI, suggesting that pitstop is blocking CIE through a site independent of clathrin. To gain further insight into how this compound might be blocking CIE, a process that occurs independently of clathrin and dynamin but is sensitive to PM cholesterol levels, we asked whether mobility of cargo proteins entering cells by CIE might be affected by pitstop 2. To do this, we labeled cells expressing SNAPTac with the non-releasable probe, Alexa 488-conjugated BG ligand, and then imaged the cells live before and after photobleaching. In control cells treated with DMSO, surface fluorescence recovered with a t1/2 of approximately 30 sec. In contrast, there was no recovery of fluorescence for the duration of the experiment in cells treated with pitstop 2, suggesting that most of the PM SNAP-Tac was immobile. This dramatic change in surface mobility was also observed for GFP-labeled H-Ras, a marker for the CIE endosomal membrane system. A similar “freezing” of the clathrin and AP2 coat complexes with pitstop 2 was also observed in the original characterization of the compound, suggesting a striking target at the PM that may cause an inhibitory effect for most endocytic events or a general global alteration of PM structure. On the other hand, we did observe that endocytosis of shiga toxin still occurred in cells treated with pitstop 2as was previously reported, although the amount of shiga toxin internalized was less than in controls. Shiga toxin may be more resistant to pitstop as compared to other endogenous CIE cargo proteins due to its ability to bind to and cluster Gb3 glycolipid, forming a tubular invaginated entry structure into cells. Taken together, our findings demonstrate that pitstop 2 cannot be used to determine that a protein enters cells by CDE since it blocks CIE as effectively as CDE. This effect, observed for many endogenous cargo proteins and in all human cell lines examined, is due to a second site of action for the compound since it still inhibits CIE in cells where clathrin has been depleted. This second site of action may explain some of the unusual behavior of cells treated with pitstop as pointed out by Lemmon and Traub. It provides a cautionary tale for the in vivo application of “specific” small molecule inhibitors developed through chemical design as this approach cannot exclude second sites of action in living cells. Improving therapy for patients with Glioblastoma multiformeis one of the biggest challenges in oncology. Although molecular targeting has shown success in many cancers, targeted therapy for GBM has yet to demonstrate an appreciable clinical survival benefit. For example, targeting of Epidermal Growth Factor Receptorwith small molecules or monoclonal antibodies has been reported to offer no survival benefit, despite the fact that EGFR is the most common genomically altered oncogene in GBM, and targeting EGFR has shown benefit in other cancers. So an important question is: can targeted therapy provide a benefit to GBM patients?
to the m2 and clathrin heavy chain depleted cells still led to a block in endocytosis of MHCI, suggesting that pitstop is blocking CIE through a site independent of clathrin. To gain further insight into how this compound might be blocking CIE, a process that occurs independently of clathrin and dynamin but is sensitive to PM cholesterol levels, we asked whether mobility of cargo proteins entering cells by CIE might be affected by pitstop 2. To do this, we labeled cells expressing SNAPTac with the non-releasable probe, Alexa 488-conjugated BG ligand, and then imaged the cells live before and after photobleaching. In control cells treated with DMSO, surface fluorescence recovered with a t1/2 of approximately 30 sec. In contrast, there was no recovery of fluorescence for the duration of the experiment in cells treated with pitstop 2, suggesting that most of the PM SNAP-Tac was immobile. This dramatic change in surface mobility was also observed for GFP-labeled H-Ras, a marker for the CIE endosomal membrane system. A similar “freezing” of the clathrin and AP2 coat complexes with pitstop 2 was also observed in the original characterization of the compound, suggesting a striking target at the PM that may cause an inhibitory effect for most endocytic events or a general global alteration of PM structure. On the other hand, we did observe that endocytosis of shiga toxin still occurred in cells treated with pitstop 2as was previously reported, although the amount of shiga toxin internalized was less than in controls. Shiga toxin may be more resistant to pitstop as compared to other endogenous CIE cargo proteins due to its ability to bind to and cluster Gb3 glycolipid, forming a tubular invaginated entry structure into cells. Taken together, our findings demonstrate that pitstop 2 cannot be used to determine that a protein enters cells by CDE since it blocks CIE as effectively as CDE. This effect, observed for many endogenous cargo proteins and in all human cell lines examined, is due to a second site of action for the compound since it still inhibits CIE in cells where clathrin has been depleted. This second site of action may explain some of the unusual behavior of cells treated with pitstop as pointed out by Lemmon and Traub. It provides a cautionary tale for the in vivo application of “specific” small molecule inhibitors developed through chemical design as this approach cannot exclude second sites of action in living cells. Improving therapy for patients with Glioblastoma multiformeis one of the biggest challenges in oncology. Although molecular targeting has shown success in many cancers, targeted therapy for GBM has yet to demonstrate an appreciable clinical survival benefit. For example, targeting of Epidermal Growth Factor Receptorwith small molecules or monoclonal antibodies has been reported to offer no survival benefit, despite the fact that EGFR is the most common genomically altered oncogene in GBM, and targeting EGFR has shown benefit in other cancers. So an important question is: can targeted therapy provide a benefit to GBM patients?
Gal-3 is expressed at much higher levels in macrophages than other cell types neural cell adhesion molecules
However, as outlined in the Introduction, other investigators have found the opposite effect in gal-3 null mice with NASH in aging mice or induced by the CDAA diet. While there is no obvious explanation for the different findings of these two groups, the preponderance of data from other gal-3 null mouse studies on fibrosis in liver, kidney, lung, and heartdemonstrates that gal-3 appears to be integral to accumulation of fibrosis in INCB18424 parenchymal tissue. In addition, while  the use of gal-3 null mice is a powerful biological model, there are some drawbacks including the obvious potential impact of background mouse strain. Also, both intracellular and extracellular gal-3 is eliminated in the null mice. The extracellular effects of galectins are related to their lectin properties to bind to glycoproteins whereas their intracellular effects are related to protein-protein interactions. Treatment with GR-MD-02, a complex carbohydrate with galactose residues would be expected to interfere with lectin effects predominantly on the cell surface and in the extracellular space. While the degree of collagen deposition in the model of NASH used in these studies is modest, as in many animal models of NASH, we have previously reported evidence that the same drug agents are effective in reducing much greater degrees of fibrosis and cirrhosis in thioacetamide-treated rats. In these previous studies, the regression in cirrhosis was associated with a reduction in portal hypertension, demonstrating that the change in liver architecture has a physiological effect on liver blood flow and/or resistance. Therefore, it appears that these drugs have effects on the early pathophysiological collagen deposition in NASH, ncluding peri-central and peri-sinusoidal deposition of collagen and potentially later stages of fibrosis and cirrhosis. The presumed proximate mechanism of action of the drugs used in this study is related to gal-3 binding. Gal-3 has a carbohydrate recognition domainwhich is shared among galectin proteins, but in contrast to other galectin proteins, it has a long N-terminal domain that is involved in forming multimers. Gal-3 binds poorly to single galactose molecules, more avidly to galactose containing disaccharides, and most avidly to larger molecules such as glycoproteins with galactose residues. We have shown that our carbohydrate drugs bind to the gal-3 CRD through somewhat different sets of amino acid residues and the affinity at 50% saturation of GR-MD02 and GM-CT-01 to gal-3 is 2.9 mM and 2.8 mM, respectively. This compares to gal-1 binding affinities for GR-MD-02 and GM-CT-01 of 8 mM and 10 mM, respectively. Although galectins are defined by their ability to bind to model carbohydrates containing galactose, such as N-acetyllactosamine, individual galectins appear to bind to different sets of glycans on glycoproteins, thus providing specificity between galectins. For example, galectin-1 and galectin-3 bind to distinct cell surface receptors on T-cells. There are many reported potential ligands for the lectin properties of galectin-3 including laminin, U0126 integrins, collagens, fibronectin, elastin, mucins, CD4+, CD8+, TGFBR, and many others. Binding of galectin-3 to Nglycans has been connected to multiple cellular processes including cell adhesion and migration, immune cell function, inflammation, and neoplasia. It is likely, that inhibition of galectin-3 modulates multiple protein interactions in the extracellular space thereby altering cellular function. We have not determined in these studies which gal-3 protein interactions are abrogated by drug treatment. However, we have data that suggest some downstream processes that are affected, one of which seems to involve macrophages.
the use of gal-3 null mice is a powerful biological model, there are some drawbacks including the obvious potential impact of background mouse strain. Also, both intracellular and extracellular gal-3 is eliminated in the null mice. The extracellular effects of galectins are related to their lectin properties to bind to glycoproteins whereas their intracellular effects are related to protein-protein interactions. Treatment with GR-MD-02, a complex carbohydrate with galactose residues would be expected to interfere with lectin effects predominantly on the cell surface and in the extracellular space. While the degree of collagen deposition in the model of NASH used in these studies is modest, as in many animal models of NASH, we have previously reported evidence that the same drug agents are effective in reducing much greater degrees of fibrosis and cirrhosis in thioacetamide-treated rats. In these previous studies, the regression in cirrhosis was associated with a reduction in portal hypertension, demonstrating that the change in liver architecture has a physiological effect on liver blood flow and/or resistance. Therefore, it appears that these drugs have effects on the early pathophysiological collagen deposition in NASH, ncluding peri-central and peri-sinusoidal deposition of collagen and potentially later stages of fibrosis and cirrhosis. The presumed proximate mechanism of action of the drugs used in this study is related to gal-3 binding. Gal-3 has a carbohydrate recognition domainwhich is shared among galectin proteins, but in contrast to other galectin proteins, it has a long N-terminal domain that is involved in forming multimers. Gal-3 binds poorly to single galactose molecules, more avidly to galactose containing disaccharides, and most avidly to larger molecules such as glycoproteins with galactose residues. We have shown that our carbohydrate drugs bind to the gal-3 CRD through somewhat different sets of amino acid residues and the affinity at 50% saturation of GR-MD02 and GM-CT-01 to gal-3 is 2.9 mM and 2.8 mM, respectively. This compares to gal-1 binding affinities for GR-MD-02 and GM-CT-01 of 8 mM and 10 mM, respectively. Although galectins are defined by their ability to bind to model carbohydrates containing galactose, such as N-acetyllactosamine, individual galectins appear to bind to different sets of glycans on glycoproteins, thus providing specificity between galectins. For example, galectin-1 and galectin-3 bind to distinct cell surface receptors on T-cells. There are many reported potential ligands for the lectin properties of galectin-3 including laminin, U0126 integrins, collagens, fibronectin, elastin, mucins, CD4+, CD8+, TGFBR, and many others. Binding of galectin-3 to Nglycans has been connected to multiple cellular processes including cell adhesion and migration, immune cell function, inflammation, and neoplasia. It is likely, that inhibition of galectin-3 modulates multiple protein interactions in the extracellular space thereby altering cellular function. We have not determined in these studies which gal-3 protein interactions are abrogated by drug treatment. However, we have data that suggest some downstream processes that are affected, one of which seems to involve macrophages.