Our drug development scheme should be applicable to the design of small molecules capable of specifically interfering with many other well-characterized inter or intra-molecular interactions with amenable surfaces. Other non-peptide small molecules that LY2109761 disrupt specific protein-protein interactions have been successfully developed in recent times, and they are beginning to show great promise for the therapy of human cancer. In practical terms, we have developed small molecules, and we have shown that these compounds can be used in combination therapy schemes as antitumoral agents in cultured and animal models of cancer. Specifically, compound Ia sensitizes PC-3 prostate cancer cells to the antiproliferative activity of doxorubicin in cultured cells and it shows direct antitumoral activity in mouse tumor xenografts. A number of mechanisms can be at play to cause increased sensitivities of tumor cells to chemotherapy or radiotherapy, including inhibition of NF-kB, downregulation of transporters of the MDR family or the Akt-mTOR pathway. The evidence provided here suggests that at least two mechanisms may be relevant for the increased sensitivity to doxorubicin caused by compound Ia, namely inhibition of NFk-B activity and compromise of DNA repair. The demonstration that this compound disrupts the interaction between Uev1 and Ubc13 provides a mechanistic explanation for its inhibitory activity on the NF-kB signaling pathway. Recently, it has been shown that another ubiquitin conjugating enzyme, UbcH5, can promote K63 polyubiquitylation, and that NF-kB activation by IL-1b is much more strongly dependent on Ubc13-dependent K63 polyubiquitylation than activation by TNF-a. However, a large body of literature strongly suggests a critical role of Ubc13 and K63 polyubiquitylation in the activation of NF-kB not only by IL-1b but also by TNF-a. In this regard, the chain type of ligand-induced ubiquitylation by cIAP of TNF-R1 complex components has not been determined, and, given the recruitment of Ubc13 by cIAP, it is quite possible that 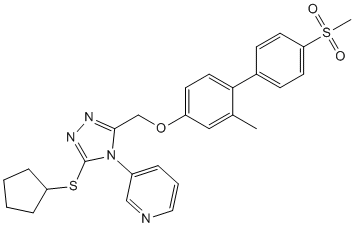 such chains are of the K63 type. Furthermore, mice haploinsuficient for Ubc13 display cell-typespecific defects in chemokine and NF-kB signaling, supporting a critical role of Ubc13 and K63 polyubiquitylation in the activation of NF-kB by different stimuli in vivo, including TNF-a and LPS. Our observations showing that the small molecule antagonist of Ubc13-Uev interactions compound Ia inhibits NF-kB activation by TNF-a would also support a role for Ubc13 in this pathway. Alternative explanations would include the possibility that our compounds inhibit other ubiquitin conjugating enzymes or additional components of the TNF-a signaling BMN673 cascade, which has not been formally ruled out in the present study. On the other hand, it has also been shown that unanchored K63-linked polyubiquitin chains are essential for the activation of the RIG-I pathway in response to viral infection, and that both Ubc13 and Ubc5 are required in this pathway. Therefore, the inhibition of Ubc13 by small compounds could limit the response to viral infections mediated through this pathway. Regarding the role of Ubc13 and K63 polyubiquitylation in DNA damage response, the very high similarity of Uev2 to Uev1, and the computed interaction of compound Ia on the hydrophobic pocket of Ubc13, allows to predict with sufficient confidence that this compound should disrupt also the interaction of Uev2 with Ubc13.
such chains are of the K63 type. Furthermore, mice haploinsuficient for Ubc13 display cell-typespecific defects in chemokine and NF-kB signaling, supporting a critical role of Ubc13 and K63 polyubiquitylation in the activation of NF-kB by different stimuli in vivo, including TNF-a and LPS. Our observations showing that the small molecule antagonist of Ubc13-Uev interactions compound Ia inhibits NF-kB activation by TNF-a would also support a role for Ubc13 in this pathway. Alternative explanations would include the possibility that our compounds inhibit other ubiquitin conjugating enzymes or additional components of the TNF-a signaling BMN673 cascade, which has not been formally ruled out in the present study. On the other hand, it has also been shown that unanchored K63-linked polyubiquitin chains are essential for the activation of the RIG-I pathway in response to viral infection, and that both Ubc13 and Ubc5 are required in this pathway. Therefore, the inhibition of Ubc13 by small compounds could limit the response to viral infections mediated through this pathway. Regarding the role of Ubc13 and K63 polyubiquitylation in DNA damage response, the very high similarity of Uev2 to Uev1, and the computed interaction of compound Ia on the hydrophobic pocket of Ubc13, allows to predict with sufficient confidence that this compound should disrupt also the interaction of Uev2 with Ubc13.
These models reveal that the unique cysteine residue is located at the entrance of the AChE
The almost negligible values of the binding constants suggest a possible lack of hormonal activity. This has been further confirmed by preclinical animal studies using a TTRV30M transgenic mice strain receiving 2.8 mg of iododiflunisal per day during 3 months. The animals did not show significant metabolic disfunctions. However, further preclinical tests are needed to validate these compounds as potential drugs for TTR related amyloidosis. In conclusion, by mimicking the natural interactions between thyroid hormones and TTR and by using diflunisal as a model compound, the biochemical and biophysical data above discussed supports the hypothesis that iodine atoms inserted in TTR binding compounds is a crucial factor for the design of novel PB 203580 152121-47-6 highly potent TTR fibrillogenesis inhibitors that one day become effective drugs for the treatment of TTR-related amyloidosis. Aphids are among the world’s most destructive insect pests of grain crops, vegetables, ornamental plants, and fruit trees. For 150 years the greenbug aphid has been a major pest of small grains. Annual costs for greenbug control in wheat production have been estimated at up to $100 million on the Texas High Plains alone. The soybean aphid causes combined US yield losses and increased production costs that exceed $1 billion. Aphid control relies mainly on a small number of highly toxic anticholinesterases approved by the US Environmental Protection Agency; the threat to agriculture and environmental health is growing. This phenomenon is partly due to an unusual feature of aphid biology. During the aphid-growing season all aphids become female, and are able to produce offspring by maternal cloning in a process known as parthenogenesis. This form of reproduction, with up to 18 asexual generations per growing season, allows aphids to develop resistance rapidly when few effective insecticides are applied repeatedly, as often happens in crops such as soybeans. Acetylcholinesterase is a serine hydrolase vital for regulating the neurotransmitter acetylcholine in mammals, birds, and insects. Current anticholinesterase insecticides such as chlorpyrifos and methamidophos phosphorylate a serine residue at the active site of AChE, thus disabling its function and causing incapacitation. Because this serine residue is also present in mammalian and avian AChEs, use of these insecticides poses serious risks of toxicity to mammals, birds, and beneficial insects such as the honeybee. The US EPA has concluded that such agents can enter the brain of fetuses and young children and may damage the developing nervous system. Controlling aphids in a large field requires insecticides at quantities toxic to mammals and birds. Unintended environmental toxicity is a concern associated with current agents used to Enzalutamide manage these insects. In light of this concern, and the problem of insecticide resistance described above, there is an urgent need for new agents that are both safer and more effective in controlling aphids and related pests. A new concept for insect control is to use an irreversible inhibitor that targets an insect-specific region of an essential protein in the target species. 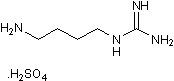 Sequence analyses of various insect proteins identified a cysteine residue that is absent in mammalian and avian AChEs but conserved in the AChEs of aphids and several other insects. This sequence-based finding was consistent with the reports that aphid AChEs were sensitive to sulfhydryl inhibitors. The sequence analysis along with the site-directed mutagenesis and molecular modeling studies on an AChE from amphioxus led to speculations that the cysteine residue conserved in the aphid AChE is located near the top of the active-site gorge and sensitive to sulfhydryl inhibitors and that high affinity bi-functional cholinergic reagents that react transiently with the active site serine and irreversibly with the cysteine residue could be candidates for selective aphicides. The threedimensional models of AChEs in the greenbug and the English grain aphid generated by using terascale computing were reported subsequently.
Sequence analyses of various insect proteins identified a cysteine residue that is absent in mammalian and avian AChEs but conserved in the AChEs of aphids and several other insects. This sequence-based finding was consistent with the reports that aphid AChEs were sensitive to sulfhydryl inhibitors. The sequence analysis along with the site-directed mutagenesis and molecular modeling studies on an AChE from amphioxus led to speculations that the cysteine residue conserved in the aphid AChE is located near the top of the active-site gorge and sensitive to sulfhydryl inhibitors and that high affinity bi-functional cholinergic reagents that react transiently with the active site serine and irreversibly with the cysteine residue could be candidates for selective aphicides. The threedimensional models of AChEs in the greenbug and the English grain aphid generated by using terascale computing were reported subsequently.
Activity of the APEC CH2 ivy knock-out could indicate that this strain also has an additional pliC
In conclusion, this work is the first to demonstrate the involvement of a lysozyme inhibitor in bacterial virulence. Although findings from the APEC – chicken model system studied in this work cannot be simply extrapolated to other pathogen – host interactions, the wide distribution of different types of lysozyme inhibitors in bacteria suggests that these molecules have evolved as virulence factors or effectors of commensal interactions in a wide range of bacteria. This finding may also open Temozolomide perspectives for new avenues for the development of antibacterial drugs, for example by designing compounds that can neutralize bacterial lysozyme inhibitors, thus rendering them more sensitive to the host lysozymes. Hepatic fibrosis is a reversible wound-healing response characterized by the accumulation of extracellular matrix in response to acute or chronic liver injury. Perpetuation of the fibrotic reaction can lead to end-stage liver disease, cirrhosis, and hepatocellular carcinoma, whose incidence is increasing worldwide. The activation and proliferation of hepatic stellate cells has been identified as a critical event in the development of hepatic fibrosis. Activated HSCs are highly contractile and express a-smooth muscle actin and ECM. They are a key target for anti-fibrotic therapies because these cells are the primary source of ECM in injured livers. The Notch signaling pathway is a highly conserved signal transduction mechanism. It is essential to normal embryonic development, cellular proliferation, specification, and differentiation. Four Notch receptors and five ligands have been identified in mammals. Notch signaling is activated through an interaction of a Notch receptor with a ligand expressed on adjacent cells leading to proteolytic cleavages of Notch receptor. The cleavage step catalyzed by the 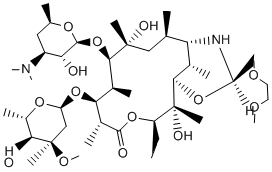 csecretase complex results in the release of the Notch intracellular domain. The NICD then moves to the nucleus, where it interacts with CSL and Mastermind to activate transcription of downstream target genes such as Hes1, HRT, Deltes-1, Meltrin-b, and the Notch receptors themselves. Notch signaling is essential to the regulation of cell differentiation, and aberrant activation of this pathway is implicated in the pathogenesis of several malignancies. Increasing numbers of studies have reported that Notch signaling is involved in human fibrotic diseases. However, the role of Notch signaling in liver fibrosis has not been fully investigated. Previous studies have indicated that all 4 receptors are expressed in the adult liver, with no significant differences in the levels of Notch1, 2, and 4 mRNA between normal and Y-27632 dihydrochloride diseased livers. However, the expression of Notch3 and Jagged1 protein has been found to be significantly up-regulated in diseased liver tissue. Recent research has found the mRNA of Notch receptors to be present in freshly isolated rat HSCs, which displayed no protein synthesis of Notch ligands. However, the amount of Jagged1 protein increased while isolated HSCs developed into myofibroblast-like cells. Based on these studies, we hypothesize that Notch signaling might be involved in liver fibrogenesis. In the present study, we investigated the role of Notch signaling during the process of liver fibrosis and clarified its mechanism. Our results demonstrated that Notch signaling is activated in hepatic fibrosis induced by CCl4 and that blocking Notch signaling using c-secretase inhibitor can significantly attenuate liver fibrosis. These results suggest that selective interruption of Notch signaling might be a novel anti-fibrotic strategy in hepatic fibrosis. In this study, we show that Notch signaling is markedly activated in a rat model of liver fibrosis induced by CCl4.
csecretase complex results in the release of the Notch intracellular domain. The NICD then moves to the nucleus, where it interacts with CSL and Mastermind to activate transcription of downstream target genes such as Hes1, HRT, Deltes-1, Meltrin-b, and the Notch receptors themselves. Notch signaling is essential to the regulation of cell differentiation, and aberrant activation of this pathway is implicated in the pathogenesis of several malignancies. Increasing numbers of studies have reported that Notch signaling is involved in human fibrotic diseases. However, the role of Notch signaling in liver fibrosis has not been fully investigated. Previous studies have indicated that all 4 receptors are expressed in the adult liver, with no significant differences in the levels of Notch1, 2, and 4 mRNA between normal and Y-27632 dihydrochloride diseased livers. However, the expression of Notch3 and Jagged1 protein has been found to be significantly up-regulated in diseased liver tissue. Recent research has found the mRNA of Notch receptors to be present in freshly isolated rat HSCs, which displayed no protein synthesis of Notch ligands. However, the amount of Jagged1 protein increased while isolated HSCs developed into myofibroblast-like cells. Based on these studies, we hypothesize that Notch signaling might be involved in liver fibrogenesis. In the present study, we investigated the role of Notch signaling during the process of liver fibrosis and clarified its mechanism. Our results demonstrated that Notch signaling is activated in hepatic fibrosis induced by CCl4 and that blocking Notch signaling using c-secretase inhibitor can significantly attenuate liver fibrosis. These results suggest that selective interruption of Notch signaling might be a novel anti-fibrotic strategy in hepatic fibrosis. In this study, we show that Notch signaling is markedly activated in a rat model of liver fibrosis induced by CCl4.
Genome sequences that were added to the NCBI genome database during the preparation of our manuscript
Single knock-outs of ivy, mliC and pliG as well as an ivy/mliC double knock-out were successfully constructed in APEC CH2, and plasmid-based complementation of the mutants with the corresponding genes was accomplished. First we determined the serum resistance of the mutants as a rapid and simple indicator of virulence, and found that mliC, but not ivy or pliG, was required for serum resistance of APEC CH2. Although bacterial sensitivity to serum is mainly due to the action of the complement system, there is also a contribution of other antimicrobial components such as lysozyme. The action of the membrane attack complex of the complement system destabilizes the outer membrane and may render it permeable to lysozyme. Conversely, degradation of the peptidoglycan layer may facilitate pore formation in the cytoplasmic membrane by the membrane attack complex, resulting in cell leakage an death. ubsequently, infection experiments with 1-day old chickens subcutaneously injected with different doses of bacteria confirmed the attenuated virulence of the mliC mutant. In addition, virulence was fully restored by complementation with the mliC gene. As BEZ235 915019-65-7 anticipated from the serum resistance test, pliG nor ivy had any significant effect on virulence. Since PliG is the only known inhibitor of g-type lysozyme in APEC, and its knock-out reduced g-type lysozyme inhibitory XAV939 284028-89-3 activity of APEC CH2 to background levels, it can be concluded that PliG is not required for virulence of this pathogen, at least not in the subcutaneous infection model used in this work. Of course, a role of this inhibitor in other commensal or pathogenic bacteria – host interactions can not be excluded on the basis of these observations. For the c-type lysozyme inhibitors, the situation is more complex. Based on the observations with the single knock-out strains, the outer membrane-bound inhibitor MliC appears to play a role in virulence, but not the periplasmic inhibitor Ivy. Since MliC is an outer membrane protein, there could be some concern that knock-out of MliC could have destabilized the outer membrane, thus rendering the bacteria more sensitive to a variety of antibacterial effectors from its host. This appears not to be the case, because the mliC mutant retained its resistance to detergents when plated on LB containing 2.0% SDS or 2.0% Triton X-100, whereas mutants with outer membrane defects typically display a high serum and detergent sensitivity. Therefore, we can have confidence that the attenuated virulence of the mliC mutant is genuinely linked to its reduced production of c-type lysozyme inhibitor rather than to an indirect effect. One point that needs further clarification is which 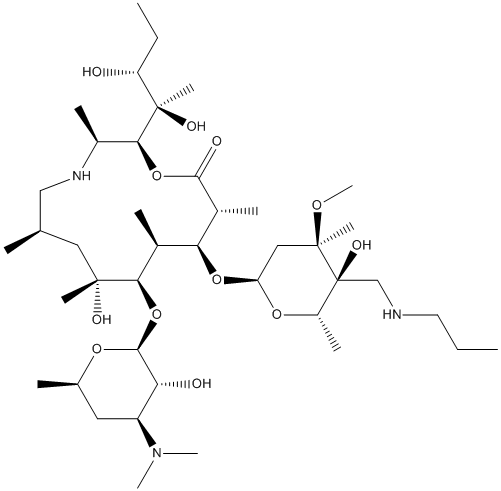 inhibitor is responsible for the attenuated virulence, since the mliC mutant unexpectedly showed a considerably reduced level of periplasmic lysozyme inhibitor activity. An additional complication, in line with the observations in the serum resistance test, is that introduction of an ivy knock-out into the mliC mutant restored the attenuated virulence of the latter to almost wild-type level again, indicating that there is some type of interference between these two mutations. Comparison of the periplasmic lysozyme inhibitory activities confirms that this is indeed the case, because the level in the double mutant is higher than that in the mliC mutant. For comparison, an ivy mliC mutant of E. coli MG1655 was previously shown to have no residual periplasmic lysozyme inhibitory activity, but an explanation for this strain-dependent behaviour is currently lacking.
inhibitor is responsible for the attenuated virulence, since the mliC mutant unexpectedly showed a considerably reduced level of periplasmic lysozyme inhibitor activity. An additional complication, in line with the observations in the serum resistance test, is that introduction of an ivy knock-out into the mliC mutant restored the attenuated virulence of the latter to almost wild-type level again, indicating that there is some type of interference between these two mutations. Comparison of the periplasmic lysozyme inhibitory activities confirms that this is indeed the case, because the level in the double mutant is higher than that in the mliC mutant. For comparison, an ivy mliC mutant of E. coli MG1655 was previously shown to have no residual periplasmic lysozyme inhibitory activity, but an explanation for this strain-dependent behaviour is currently lacking.
Viability was reduced less in the wild type cell lines to benzothiazole-based compounds with favourable biological activities
Correlating their inhibitory potencies with the pharmacophore model gave information about probable binding modes. Ocular melanomas represent approximately 5% of all melanomas, with a majority of these being uveal in origin. Uveal melanoma is the most common primary intraocular malignant tumor in adults, with an annual incidence of seven cases per million. Approximately 50% of UM patients develop metastatic melanoma to the liver within 15 years of initial diagnosis. With distant metastases, there currently is no effective treatment modality. The median survival for UM patients with metastasis is less than six months. The etiology of UM has not been fully understood. Although uveal and cutaneous melanomas arise from the same cell type, they have distinct genetic alterations. Genetic mutations in the TP53, BRAF, RAS, CDKN2 and PTEN genes are common in cutaneous melanoma but rare in UM. Drugs commonly used to treat cutaneous melanoma seldom produce durable responses in UM patients. The preponderance of liver metastases in uveal melanoma patients has focused therapeutic effort in local control of metastatic disease for palliation. Recently, somatic mutations in the GNAQ gene have been identified in about 50% of UM and 83% blue naevi. GNAQ mutations occurring at codon 209 of the RAS-like domain result in constitutive activation of the MAPK/Erk1/2 pathway in melanocytes and confer dominantly acting oncogenic functions to GNAQ. The GNAQ gene encodes for the a subunit of q class of heterotrimeric GTP binding protein that mediates signals from G-protein-coupled receptors and stimulates all four isoforms of b phospholipase C. PLCb enzymes catalyze the hydrolysis of phosphatidylinositol biphosphate, to release inositol trisphosphate and diacylglycerol that function as second messengers and propagate and amplify the Ga-mediated signal through stimulation of protein kinase C. It has been hypothesized that signaling from GNAQ to MAPK/Erk1/2 is transmitted through DAG/ PKC. The PKC family is a widely expressed group of serine/ threonine kinases comprising at least twelve isoforms. PKCs are involved in key cellular processes including cell proliferation, apoptosis, and differentiation. Increased PKC expression and activity have been demonstrated in many cancers. PKCs may play important roles in tumor formation and  progression, invasiveness of cancer cells, and chemoresistance. The mechanisms by which PKCs contribute to tumorigenesis, however, are not fully understood. Enzastaurin is a potent and selective competitive inhibitor of PKCb at low concentrations and AZD6244 inhibits other PKC isoenzymes at ALK5 Inhibitor II higher concentrations. In addition, enzastaurin targets the phosphatidylinositol 3-kinase/ AKT pathway, and inhibits phosphorylation of GSK3b and ribosomal protein S6. Although enzastaurin was initially developed as an antiangiogenic agent, it also has direct proapoptotic and antiproliferative activities on various human cancer cells. Therefore, enzastaurin may exhibit antitumor activity through multiple mechanisms affecting both tumor angiogenesis and apoptosis. Given the importance of PKC in tumorigenesis and potentially in GNAQ mutation-induced MAPK activation, we hypothesized that PKC may provide new opportunities for therapeutic intervention of UM carrying GNAQ mutations. In the present study, we tested this hypothesis by examining the response of UM cells with wild type or mutant GNAQ toward the antiproliferative and proapoptotic action of enzastaurin and characterized the underlying signaling and molecular mechanisms. The statistical model results indicate that the effect of enzastaurin upon viability depends upon both the mutational status and enzastaurin concentration.
progression, invasiveness of cancer cells, and chemoresistance. The mechanisms by which PKCs contribute to tumorigenesis, however, are not fully understood. Enzastaurin is a potent and selective competitive inhibitor of PKCb at low concentrations and AZD6244 inhibits other PKC isoenzymes at ALK5 Inhibitor II higher concentrations. In addition, enzastaurin targets the phosphatidylinositol 3-kinase/ AKT pathway, and inhibits phosphorylation of GSK3b and ribosomal protein S6. Although enzastaurin was initially developed as an antiangiogenic agent, it also has direct proapoptotic and antiproliferative activities on various human cancer cells. Therefore, enzastaurin may exhibit antitumor activity through multiple mechanisms affecting both tumor angiogenesis and apoptosis. Given the importance of PKC in tumorigenesis and potentially in GNAQ mutation-induced MAPK activation, we hypothesized that PKC may provide new opportunities for therapeutic intervention of UM carrying GNAQ mutations. In the present study, we tested this hypothesis by examining the response of UM cells with wild type or mutant GNAQ toward the antiproliferative and proapoptotic action of enzastaurin and characterized the underlying signaling and molecular mechanisms. The statistical model results indicate that the effect of enzastaurin upon viability depends upon both the mutational status and enzastaurin concentration.