As previously pointed out, this reflects the limits of our prior technology. As discussed previously, homozygosity of tumor oncogenes in cancer cell lines is frequent, although the available information did not permit the distinction between MASI with CNGs or UPD as the mechanism. Using gene-specific and genome-wide approaches we found that UPD was frequent for three EGFR pathway genes, especially for KRAS gene. Relatively little data exists in the literature for KRAS CNGs in human tumors. Furthermore, KRAS homozygosity was observed independent of mutational status as previously described. The wild type allele of KRAS can also inhibit lung carcinogenesis in mice, providing a possible 20S-Notoginsenoside-R2 explanation for the frequent finding of UPD with mutant and wild type oncogenes. 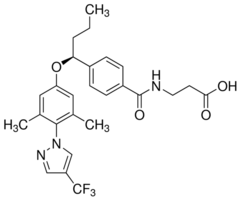 MASI has apparent biological and clinical significance. MASI at the genomic level was precisely maintained after transcription. While mutations, CNGs and allelic imbalance of mA and wA may all contribute to tumorigenesis, combinations of the three events may be more effective than any single event. Evidence for this concept was provided by our finding that the combination of mutation and CNGs acted synergistically to enhance ras GTPase activity. A recent report found that all KRAS mutations did not exert an equal effect on tumor cells. Cancer cell lines harboring KRAS mutations could be broadly divided into KRAS-dependent and KRAS-independent groups. The vast majority of KRAS-dependent lines exhibited focal KRAS CNGs, in contrast to KRAS-independent lines. This study provides further evidence that the combination of KRAS mutations and CNGs act synergistically. Our previous findings that EGFR mutations were associated with tumor initiation while EGFR CNG might be more regarded as a tumor progression event, provide further evidence of their co-operative role in tumorgenesis. Understanding the mechanism of MASI could elucidate new understandings of tumor biology and may contribute to the development of rational targeted therapies. MASI in its various forms is frequently present in mutant EGFR and KRAS tumor cells, and is associated with increased mutant allele transcription and gene activity. The frequent finding of mutations, copy number gains and MASI occurring together in tumor cells indicates that these three genetic alterations, acting together, may have a greater role in the development or maintenance of the malignant phenotype than any individual alteration. Microarray gene expression data have been integrated to increase statistical power. Increasing sample size is a bottleneck in DNA microarray-based gene expression studies as microarray experiments are time consuming, expensive, noisy and limited to the number of biological samples. To circumvent this problem, microarray gene expression data sets addressing the same or similar biological questions have been analyzed jointly either by so-called meta analysis, which means integration at the level of results derived separately from individual data sets, or by data 4-(Benzyloxy)phenol merging. But data integration prior to analysis potentially faces problems related to reproducibility as different microarray platforms use different probes for the same genes and return expression values on different numerical scales. Some studies combined data sets generated with the same chip, or with the same technology platform but different chips, or with heterogeneous microarray technologies. The rationale behind combining data sets generated only from the same chip or platform was to avoid the cross-platform bias.
MASI has apparent biological and clinical significance. MASI at the genomic level was precisely maintained after transcription. While mutations, CNGs and allelic imbalance of mA and wA may all contribute to tumorigenesis, combinations of the three events may be more effective than any single event. Evidence for this concept was provided by our finding that the combination of mutation and CNGs acted synergistically to enhance ras GTPase activity. A recent report found that all KRAS mutations did not exert an equal effect on tumor cells. Cancer cell lines harboring KRAS mutations could be broadly divided into KRAS-dependent and KRAS-independent groups. The vast majority of KRAS-dependent lines exhibited focal KRAS CNGs, in contrast to KRAS-independent lines. This study provides further evidence that the combination of KRAS mutations and CNGs act synergistically. Our previous findings that EGFR mutations were associated with tumor initiation while EGFR CNG might be more regarded as a tumor progression event, provide further evidence of their co-operative role in tumorgenesis. Understanding the mechanism of MASI could elucidate new understandings of tumor biology and may contribute to the development of rational targeted therapies. MASI in its various forms is frequently present in mutant EGFR and KRAS tumor cells, and is associated with increased mutant allele transcription and gene activity. The frequent finding of mutations, copy number gains and MASI occurring together in tumor cells indicates that these three genetic alterations, acting together, may have a greater role in the development or maintenance of the malignant phenotype than any individual alteration. Microarray gene expression data have been integrated to increase statistical power. Increasing sample size is a bottleneck in DNA microarray-based gene expression studies as microarray experiments are time consuming, expensive, noisy and limited to the number of biological samples. To circumvent this problem, microarray gene expression data sets addressing the same or similar biological questions have been analyzed jointly either by so-called meta analysis, which means integration at the level of results derived separately from individual data sets, or by data 4-(Benzyloxy)phenol merging. But data integration prior to analysis potentially faces problems related to reproducibility as different microarray platforms use different probes for the same genes and return expression values on different numerical scales. Some studies combined data sets generated with the same chip, or with the same technology platform but different chips, or with heterogeneous microarray technologies. The rationale behind combining data sets generated only from the same chip or platform was to avoid the cross-platform bias.
Whilst severity of disease is influenced by trimester in which infection is acquired including genetic predisposition may contribute
New ocular lesions can occur at any age after birth, in untreated and some treated children. For example, previous studies suggest that genes affecting immune response, including HLA, influence clinical outcome in the child. However, since infants who have the most severe clinical signs in the brain and eye are those infected early in pregnancy when fetal immunity is least well developed, we considered whether genes that encode molecules that play a role in developmental processes could contribute to clinical phenotype observed in the child. This could provide unique insight into events in utero and post-natally that determine the clinical outcome of infection. In particular, we hypothesized that propensity for T. gondii to cause eye disease may be associated with genes previously implicated in congenital or juvenile onset ocular disease. Two genes were of specific interest, ABCA4 encoding ATP-binding cassette transporter subfamily A member 4 associated with juvenile onset retinal dystrophies including Stargardt��s disease, and COL2A1 encoding type II collagen associated with Stickler syndrome in which there is congenital abnormal vitreous and lattice retinal degeneration. Although risk of transmission of the parasite to the fetus in pregnant women with primary infection ranges from,1�C 100% depending on the time in gestation when infection is acquired, incidence rates of clinical congenital toxoplasmosis, together with the fact that it is not a reportable disease, have 20S-Notoginsenoside-R2 precluded accumulation of large cohorts for genetic studies. Nevertheless, two unique cohorts have recently become available to test this specific genetic hypothesis. These cohorts are from the European Multicentre Cohort Study on Congenital Toxoplasmosis which recruited prospectively for mothers with primary T. gondii in pregnancy, and from the National Collaborative Chicago-based Congenital Toxoplasmosis Study in North America to which infants and children with congenital infection with T. gondii are referred. Using these cohorts we show that ocular and brain disease in congenital toxoplasmosis associate with polymorphisms in ABCA4, while polymorphisms at COL2A1 encoding type II collagen associate only with ocular disease. Both loci show unusual inheritance patterns for the disease allele when comparing outcomes in heterozygous affected children with outcomes in affected children of heterozygous mothers, and modeling suggests either an effect of mother��s genotype or parent-of-origin effects. The latter is supported by experimental data showing that both ABCA4 and COL2A1 show isoform-specific epigenetic modifications consistent with imprinting. 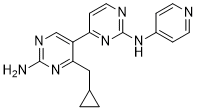 The European study was undertaken as an adjunct to European Multicentre Study of Congenital Toxoplasmosis, referred to as GENET-EMSCOT. Ethical approvals for GENET-EMSCOT were obtained through the local ethical review boards of participating centres across Europe, and for the study as a whole from the Research Ethics Committee for Great Ormond Street Hospital and the Institute for Child Health in London. Screening, treatment and follow up schedules have been described in detail elsewhere. For this study, mother-child pairs were eligible for inclusion if the child had congenital toxoplasmosis or, if the child was uninfected, the mother had evidence of seroconversion during pregnancy. Infected children were identified in centres that utilized universal prenatal or neonatal screening. Uninfected children were identified only in those centres that utilized prenatal screening. Families were Benzethonium Chloride invited to participate by the clinician responsible for follow up.
The European study was undertaken as an adjunct to European Multicentre Study of Congenital Toxoplasmosis, referred to as GENET-EMSCOT. Ethical approvals for GENET-EMSCOT were obtained through the local ethical review boards of participating centres across Europe, and for the study as a whole from the Research Ethics Committee for Great Ormond Street Hospital and the Institute for Child Health in London. Screening, treatment and follow up schedules have been described in detail elsewhere. For this study, mother-child pairs were eligible for inclusion if the child had congenital toxoplasmosis or, if the child was uninfected, the mother had evidence of seroconversion during pregnancy. Infected children were identified in centres that utilized universal prenatal or neonatal screening. Uninfected children were identified only in those centres that utilized prenatal screening. Families were Benzethonium Chloride invited to participate by the clinician responsible for follow up.
SMAR1 acts as a key regulator of induction of p53 acetylation corresponds with SMAR1 induction
Chromatin immunoprecipitation assays showed that Doxorubicin induced DNA damage Cinoxacin consequently leads to the recruitment of acetylated p53 on SMAR1 promoter and thus activate its transcription. Along with p53 acetylation, recruitment of p53 dependent activator complex on SMAR1 promoter also resulted in H3-K9 acetylation indicative of active chromatin at the locus. Once SMAR1 reaches a threshold expression, it allows deacetylation of p53 as shown upon SMAR1 overexpression. Upregulation of acetylated p53 is followed by induction in SMAR1 expression that is further correlated with the cell cycle where the SMAR1 expression was highest during G1/S phase. Prolonged treatment of Doxorubicin resulted in G2/M arrest, which can be explained due to SMAR1 mediated stabilization of p53 in the nucleus. siRNA treatment of SMAR1 inhibited Doxorubicin mediated cell cycle arrest. Cell cycle progression is thus regulated by p53 in response to DNA damage through regulation of SMAR1 expression that is directly involved in chromatin remodeling at the Cyclin D1 promoter loci. This suggests that SMAR1 functions in Atractylenolide-III synergism with the Doxorubicin by conferring selective repressor activity to p53 through its deacetylation that results only in cell cycle arrest and not apoptosis. Thus, in addition to previously reported pathways here we report a new positive feedback regulation between p53 and SMAR1 where SMAR1 is transcriptionally activated by p53 and that in turn stabilize p53 by facilitating phosphorylation at serine 15 residue. The expression of SMAR1 is drastically downregulated during breast cancer progression and is inversely correlated with Cyclin D1 expression. Interestingly, we observed that acetylated p53 at K373/382 showed diffused expression in the nuclear spaces with higher expression in the nucleolar compartments in breast fibroadenoma 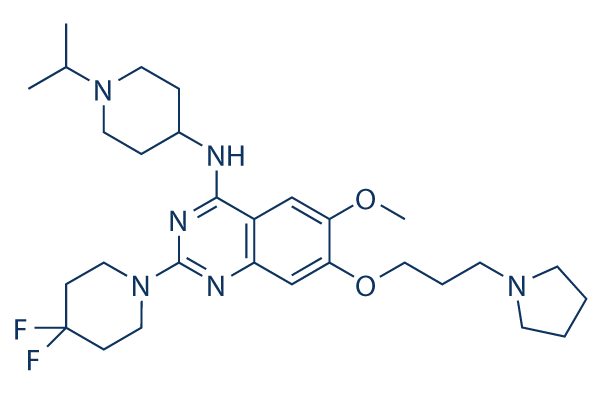 and exclusively nucleolar translocation in IDC G-III samples. These observations suggest that mutations in p53 may lead to its altered status and expression pattern in human breast carcinoma and is associated with its sequestration into heterochromatin resulting in its compromised activity. Similar correlation between p53 localization was observed in various breast carcinoma cell lines where p53 and acetylated p53 were predominantly present in the heterochromatin regions. All above observations suggest a possible mechanism of SMAR1 dysregulation in breast cancer due to abnormal p53 acetylation, phosphorylation and its sub-cellular sequestration. Moreover, SMAR1 leads to reduced migration and invasion in both poorly and highly metastatic breast carcinoma cell lines irrespective of p53 status in both transient and stably SMAR1 transfected cells through downregulation of TGFb signaling and its target gene expression including CUTL1. Earlier we have reported that SMAR1 inhibits ERK phosphorylation that may contribute to decreased phospho-Smad2 levels and thus inhibits TGFb signaling. SMAR1 is also able to reduce metastatic potential of B16F1 cells in-vivo in mice model. Microarray analysis done in SMAR1 stable clone, showing downregulation of various oncogenes, Cyclins, focal-adhesion molecules, TGFb related genes and upregulation of various growth inhibitory, DNA repair and stress response genes also indicated anti-tumorigenic and antimetastatic function of SMAR1. Our results establish SMAR1, as a critical regulator of TGFb signaling cascade that finally affect CUTL1 expression. Downregulation of SMAR1 in high-grade breast carcinoma can be directly correlated with activated TGFb signaling and its downstream target genes that consequently lead to increased tumor metastasis.
and exclusively nucleolar translocation in IDC G-III samples. These observations suggest that mutations in p53 may lead to its altered status and expression pattern in human breast carcinoma and is associated with its sequestration into heterochromatin resulting in its compromised activity. Similar correlation between p53 localization was observed in various breast carcinoma cell lines where p53 and acetylated p53 were predominantly present in the heterochromatin regions. All above observations suggest a possible mechanism of SMAR1 dysregulation in breast cancer due to abnormal p53 acetylation, phosphorylation and its sub-cellular sequestration. Moreover, SMAR1 leads to reduced migration and invasion in both poorly and highly metastatic breast carcinoma cell lines irrespective of p53 status in both transient and stably SMAR1 transfected cells through downregulation of TGFb signaling and its target gene expression including CUTL1. Earlier we have reported that SMAR1 inhibits ERK phosphorylation that may contribute to decreased phospho-Smad2 levels and thus inhibits TGFb signaling. SMAR1 is also able to reduce metastatic potential of B16F1 cells in-vivo in mice model. Microarray analysis done in SMAR1 stable clone, showing downregulation of various oncogenes, Cyclins, focal-adhesion molecules, TGFb related genes and upregulation of various growth inhibitory, DNA repair and stress response genes also indicated anti-tumorigenic and antimetastatic function of SMAR1. Our results establish SMAR1, as a critical regulator of TGFb signaling cascade that finally affect CUTL1 expression. Downregulation of SMAR1 in high-grade breast carcinoma can be directly correlated with activated TGFb signaling and its downstream target genes that consequently lead to increased tumor metastasis.
It is possible that phosphorylation of eIF2a and inhibition of proliferation is apoptosis and has been linked
Interestingly, ER stress signaling has been shown to be a negative regulator of malignancy in human squamous carcinoma cells and of H-Ras-mediated transformation of human melanocytes. Further, inhibition of PKR and subsequent reduced 20S-Notoginsenoside-R2 phosphorylation of eIF2a was sufficient to cause transformation of mouse NIH3T3 fibroblasts. These results suggest that phosphorylation of eIF2a could potentially have a tumor inhibitory function. Our results reveal two important and previously unrecognized salient findings: the first is a link between adhesion signaling and PERK-dependent phosphorylation of eIF2a; the second, is the adhesion-dependent role of PERK in inhibiting proliferation and tumor formation both in 3D in vitro and in vivo animal models, respectively. Early studies showed that MEFs in suspension displayed translational repression. More recent studies showed that in NIH3T3 fibroblasts, the suspension-induced translation repression correlated with increased P-eIF2a levels. Our results are in accordance with, and further extend these findings by showing that loss of adhesion can regulate eIF2a phosphorylation in a PERK-dependent manner. That PERK is responsible for eIF2a phosphorylation, is supported by our results showing that PERK-/MEFs display reduced phosphorylation of eIF2a as compared to wtPERK MEFs placed in suspension. Importantly, activation or inhibition of PERK-independent pathways appeared to strongly regulate suspension-induced translational repression, as inhibition of PERK was not sufficient to restore protein synthesis and prevent anoikis. Alternatively, this could be due to residual eIF2a phosphorylation, since as little as 10% of eIF2a phosphorylation can cause a strong repression of translation. Another possibility maybe the fact that the eIF4E pathway may still be inhibited in suspension. It is also possible that phosphorylation of PERK and eIF2a in suspension may have other functions not immediately linked to apoptosis but to other forms of cell death. Moreover, it was interesting to find that PERKDC enhanced protein synthesis in adhered conditions and prevented DTT-induced translational repression. This suggests that PERK signaling may be required in the adherent cells to inhibit proliferation and PERKDC-expressing cells might be refractory to these signals during acinus formation. Perhaps, maintenance of LN-5 expression at basal levels is required to prevent hyperproliferation as only PERKDC MCF10A cells were surrounded by large LN-5 deposits and positive for Ki67 in the lumen-filled acini. The signals that activate PERK during acinar morphogenesis are unknown but it is possible that more subtle changes in adhesion or in matrix composition that activate PERK may become evident as the acinus reaches its terminal size. Our results showed that the proliferative and tumor suppressive effect of PERK and eIF2a signaling is not due to their ability to induce anoikis in response to loss of adhesion, but due to the inhibition of proliferation in adherent cells. The PERKDCinduced stimulation of proliferation and tumor initiation was Alprostadil unexpected and raises the question of the mechanisms behind this phenomenon. The PERKDC-induced phenotype resembled, although not as strongly, that of ErbB2 activation in MCF10A cells. To conclude, our studies reveal that PERK activation and possibly downstream eIF2a signaling is regulated by adhesion through an as yet unknown mechanism. This signal appears to be required to limit proliferation and allow for normal acinar 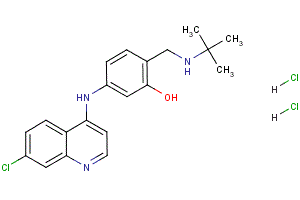 morphogenesis. Most importantly, this pathway appears to have a tumor suppressive effect.
morphogenesis. Most importantly, this pathway appears to have a tumor suppressive effect.
T cell activation and the ensuing differentiation to effector cells or is one of selective force driving clonal evolution
This view of the origin of cancer, that we refer to as a Phoenix Paradigm, has obvious implications for not only a better understanding of cancer pathogenesis, but also for the development of effective strategies for its prevention and treatment, and deserves experimental confirmation. The causes of this variability remain unclear. In somecases the validity of the definition of the species is necessary and the apparent intraspecies variation is really interspecies variation. Interspecies hybridization may be a major mechanism of diversification of the composition of snake venoms. Whatever the origin, the diversity of snake venom is important both for our understanding of venomous snake evolution and for the preparation of relevant antivenoms to treat envenomations. This was true whether these toxins acted at the presynaptic part of the neuromuscular junction, like the momomeric ammodytoxin, or bound to the postsynaptic part, like heterodimeric vaspin. We used several approaches to characterize the venom PLA2 composition of snakes captured in the areas in which the epidemiological survey was performed. We analyzed the genes and transcripts encoding venom PLA2s. We used SELDI technology to study the diversity of PLA2 neurotoxins in various venom samples. Electrospray and MALDI-MS is a faster, more accurate approach than SDS-PAGE for the characterization of venom components. SELDI-TOF-MS can be considered as an extension of the matrix-assisted laser desorption/ionization method. In the SELDI method, protein solutions are applied to the spots of ProteinChip Arrays, which have been derivatized with planar chromatographic chemistries. The proteins actively interact with the chromatographic array surface, and become sequestered as well as separated from salts and other sample contaminants by subsequent on-spot washing with appropriate buffer solutions. Prefractionation, a sample preparation prerequisite that complicates the MALDI analysis, often resulting in sample loss as well as artifactual qualitative and quantitative variances, is therefore not required for SELDI analyses. This is particularly interesting if one works with samples of which the quantities are reduced. In parallel, we evaluated venom neurotoxicity by analyzing cross-reactivity with anti-Atx antibodies. We have previously used this method to detect neurotoxins in the blood of patients bitten by vipers in the south-east of France who presented neurological symptoms. Three snake populations were identified as worthy of particular attention based on their neurotoxic venom PLA2 content. Our work demonstrates that a multidisciplinary approach can fully characterize the diversity in snake venom composition and Chloroquine Phosphate identify its medical and public health consequences. Thus, by combining epidemiological data concerning snake bites with genomic, tanscriptomic, Albaspidin-AA proteomic and immunochemical analyses of the major toxic components of venoms, we were able to provide a clear description of the potential neurotoxicity of Vipera aspis bites in France. Understanding the structure and dynamics of biological networks may prove critical to unravel complex traits and diseases, such as autoimmune diseases. In the immune response, T cells interact with antigen-presenting cells in a complex process that generates changes in gene expression. These changes underlie cell differentiation, and effector and regulatory events, as well as promoting the acquisition of a panel of adhesion molecules that guide cells to the appropriate  tissues. Several evidences indicates gene deregulation within the immune system in autoimmune diseases, such as in Multiple Sclerosis.
tissues. Several evidences indicates gene deregulation within the immune system in autoimmune diseases, such as in Multiple Sclerosis.