Single knock-outs of ivy, mliC and pliG as well as an ivy/mliC double knock-out were successfully constructed in APEC CH2, and plasmid-based complementation of the mutants with the corresponding genes was accomplished. First we determined the serum resistance of the mutants as a rapid and simple indicator of virulence, and found that mliC, but not ivy or pliG, was required for serum resistance of APEC CH2. Although bacterial sensitivity to serum is mainly due to the action of the complement system, there is also a contribution of other antimicrobial components such as lysozyme. The action of the membrane attack complex of the complement system destabilizes the outer membrane and may render it permeable to lysozyme. Conversely, degradation of the peptidoglycan layer may facilitate pore formation in the cytoplasmic membrane by the membrane attack complex, resulting in cell leakage an death. ubsequently, infection experiments with 1-day old chickens subcutaneously injected with different doses of bacteria confirmed the attenuated virulence of the mliC mutant. In addition, virulence was fully restored by complementation with the mliC gene. As BEZ235 915019-65-7 anticipated from the serum resistance test, pliG nor ivy had any significant effect on virulence. Since PliG is the only known inhibitor of g-type lysozyme in APEC, and its knock-out reduced g-type lysozyme inhibitory XAV939 284028-89-3 activity of APEC CH2 to background levels, it can be concluded that PliG is not required for virulence of this pathogen, at least not in the subcutaneous infection model used in this work. Of course, a role of this inhibitor in other commensal or pathogenic bacteria – host interactions can not be excluded on the basis of these observations. For the c-type lysozyme inhibitors, the situation is more complex. Based on the observations with the single knock-out strains, the outer membrane-bound inhibitor MliC appears to play a role in virulence, but not the periplasmic inhibitor Ivy. Since MliC is an outer membrane protein, there could be some concern that knock-out of MliC could have destabilized the outer membrane, thus rendering the bacteria more sensitive to a variety of antibacterial effectors from its host. This appears not to be the case, because the mliC mutant retained its resistance to detergents when plated on LB containing 2.0% SDS or 2.0% Triton X-100, whereas mutants with outer membrane defects typically display a high serum and detergent sensitivity. Therefore, we can have confidence that the attenuated virulence of the mliC mutant is genuinely linked to its reduced production of c-type lysozyme inhibitor rather than to an indirect effect. One point that needs further clarification is which 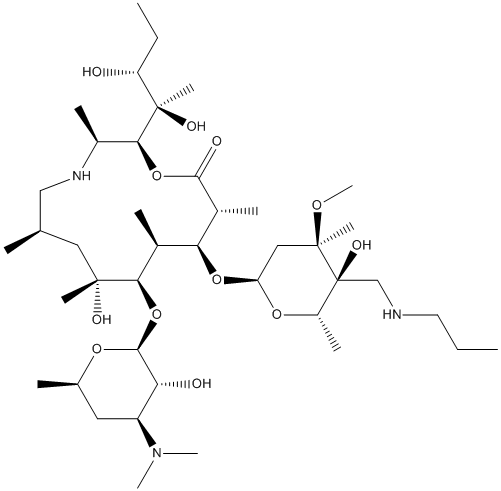 inhibitor is responsible for the attenuated virulence, since the mliC mutant unexpectedly showed a considerably reduced level of periplasmic lysozyme inhibitor activity. An additional complication, in line with the observations in the serum resistance test, is that introduction of an ivy knock-out into the mliC mutant restored the attenuated virulence of the latter to almost wild-type level again, indicating that there is some type of interference between these two mutations. Comparison of the periplasmic lysozyme inhibitory activities confirms that this is indeed the case, because the level in the double mutant is higher than that in the mliC mutant. For comparison, an ivy mliC mutant of E. coli MG1655 was previously shown to have no residual periplasmic lysozyme inhibitory activity, but an explanation for this strain-dependent behaviour is currently lacking.
inhibitor is responsible for the attenuated virulence, since the mliC mutant unexpectedly showed a considerably reduced level of periplasmic lysozyme inhibitor activity. An additional complication, in line with the observations in the serum resistance test, is that introduction of an ivy knock-out into the mliC mutant restored the attenuated virulence of the latter to almost wild-type level again, indicating that there is some type of interference between these two mutations. Comparison of the periplasmic lysozyme inhibitory activities confirms that this is indeed the case, because the level in the double mutant is higher than that in the mliC mutant. For comparison, an ivy mliC mutant of E. coli MG1655 was previously shown to have no residual periplasmic lysozyme inhibitory activity, but an explanation for this strain-dependent behaviour is currently lacking.
Month: August 2019
Viability was reduced less in the wild type cell lines to benzothiazole-based compounds with favourable biological activities
Correlating their inhibitory potencies with the pharmacophore model gave information about probable binding modes. Ocular melanomas represent approximately 5% of all melanomas, with a majority of these being uveal in origin. Uveal melanoma is the most common primary intraocular malignant tumor in adults, with an annual incidence of seven cases per million. Approximately 50% of UM patients develop metastatic melanoma to the liver within 15 years of initial diagnosis. With distant metastases, there currently is no effective treatment modality. The median survival for UM patients with metastasis is less than six months. The etiology of UM has not been fully understood. Although uveal and cutaneous melanomas arise from the same cell type, they have distinct genetic alterations. Genetic mutations in the TP53, BRAF, RAS, CDKN2 and PTEN genes are common in cutaneous melanoma but rare in UM. Drugs commonly used to treat cutaneous melanoma seldom produce durable responses in UM patients. The preponderance of liver metastases in uveal melanoma patients has focused therapeutic effort in local control of metastatic disease for palliation. Recently, somatic mutations in the GNAQ gene have been identified in about 50% of UM and 83% blue naevi. GNAQ mutations occurring at codon 209 of the RAS-like domain result in constitutive activation of the MAPK/Erk1/2 pathway in melanocytes and confer dominantly acting oncogenic functions to GNAQ. The GNAQ gene encodes for the a subunit of q class of heterotrimeric GTP binding protein that mediates signals from G-protein-coupled receptors and stimulates all four isoforms of b phospholipase C. PLCb enzymes catalyze the hydrolysis of phosphatidylinositol biphosphate, to release inositol trisphosphate and diacylglycerol that function as second messengers and propagate and amplify the Ga-mediated signal through stimulation of protein kinase C. It has been hypothesized that signaling from GNAQ to MAPK/Erk1/2 is transmitted through DAG/ PKC. The PKC family is a widely expressed group of serine/ threonine kinases comprising at least twelve isoforms. PKCs are involved in key cellular processes including cell proliferation, apoptosis, and differentiation. Increased PKC expression and activity have been demonstrated in many cancers. PKCs may play important roles in tumor formation and 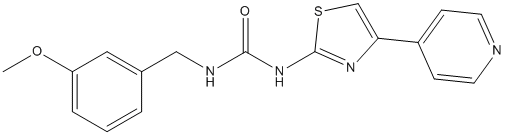 progression, invasiveness of cancer cells, and chemoresistance. The mechanisms by which PKCs contribute to tumorigenesis, however, are not fully understood. Enzastaurin is a potent and selective competitive inhibitor of PKCb at low concentrations and AZD6244 inhibits other PKC isoenzymes at ALK5 Inhibitor II higher concentrations. In addition, enzastaurin targets the phosphatidylinositol 3-kinase/ AKT pathway, and inhibits phosphorylation of GSK3b and ribosomal protein S6. Although enzastaurin was initially developed as an antiangiogenic agent, it also has direct proapoptotic and antiproliferative activities on various human cancer cells. Therefore, enzastaurin may exhibit antitumor activity through multiple mechanisms affecting both tumor angiogenesis and apoptosis. Given the importance of PKC in tumorigenesis and potentially in GNAQ mutation-induced MAPK activation, we hypothesized that PKC may provide new opportunities for therapeutic intervention of UM carrying GNAQ mutations. In the present study, we tested this hypothesis by examining the response of UM cells with wild type or mutant GNAQ toward the antiproliferative and proapoptotic action of enzastaurin and characterized the underlying signaling and molecular mechanisms. The statistical model results indicate that the effect of enzastaurin upon viability depends upon both the mutational status and enzastaurin concentration.
progression, invasiveness of cancer cells, and chemoresistance. The mechanisms by which PKCs contribute to tumorigenesis, however, are not fully understood. Enzastaurin is a potent and selective competitive inhibitor of PKCb at low concentrations and AZD6244 inhibits other PKC isoenzymes at ALK5 Inhibitor II higher concentrations. In addition, enzastaurin targets the phosphatidylinositol 3-kinase/ AKT pathway, and inhibits phosphorylation of GSK3b and ribosomal protein S6. Although enzastaurin was initially developed as an antiangiogenic agent, it also has direct proapoptotic and antiproliferative activities on various human cancer cells. Therefore, enzastaurin may exhibit antitumor activity through multiple mechanisms affecting both tumor angiogenesis and apoptosis. Given the importance of PKC in tumorigenesis and potentially in GNAQ mutation-induced MAPK activation, we hypothesized that PKC may provide new opportunities for therapeutic intervention of UM carrying GNAQ mutations. In the present study, we tested this hypothesis by examining the response of UM cells with wild type or mutant GNAQ toward the antiproliferative and proapoptotic action of enzastaurin and characterized the underlying signaling and molecular mechanisms. The statistical model results indicate that the effect of enzastaurin upon viability depends upon both the mutational status and enzastaurin concentration.
Finding an explanation for how mTOR inhibition protects against seizures could help facilitate
The most commonly used metabolism-based therapy is the high-fat, low carbohydrate ketogenic diet. The efficacy of the ketogenic diet in children was shown in a randomized controlled trial showing a AZ 960 msds robust 75% decrease in patient seizures over three months. Small molecules that potentially target the same pathways are being investigated for antiseizure effects, including agents that act on nutrient-sensing mechanisms such as the mTOR-containing TORC1 complex. In cell culture models, depletion of glucose and specific amino acids suppresses mTOR serine-threonine kinase activity, leading to reduced protein translation and induction of autophagy. Mutations in TSC1/2, genes that normally suppress mTOR, are responsible for tuberous sclerosis complex, which includes seizures, tubers, subependymal giant cell tumors, autism, behavior problems, and other systemic complications. In Tsc1- or Ptendeficient mice that have increased mTOR activity and chronic spontaneous seizures, sustained treatment with the mTOR inhibitor rapamycin decreased seizure frequency. Furthermore, the rapamycin analog everolimus restricted tumor growth and decreased seizure frequency in a clinical trial of patients with tuberous sclerosis complex. Inhibitors of mTOR may improve seizure control in other chronic epilepsy models where the underlying cause of epilepsy is not due to mutations in the TOR pathway. For example, rapamycin suppressed behavioral spasms in the doxorubicin/ lipopolysaccharide/p-chlorophenylalanine model of infantile spasms. Rapamycin also decreased susceptibility to kainic acid-induced seizures in P13 rats exposed to graded hypoxia at P10. In addition, rapamycin protected against spontaneous seizures that recur for several months following one-time kainic acid- or pilocarpine-induced status epilepticus in rats. Collectively, these reports with chronic models support the general opinion that rapamycin protects by inducing long-term cellular changes. Rapamycin also protected against seizures when administered after the initial induction of status epilepticus in the pilocarpine rat model, raising the possibility that rapamycin also may act acutely to inhibit seizure activity. However, rapamycin failed to protect when the same post-treatment model of pilocarpine-induced status epilepticus was applied to mice and it did not protect against seizures during the first 48 hours after a hypoxic insult in P10 rats, challenging the idea that rapamycin has acute antiseizure effects. Similarly, attempts to study the short-term Niltubacin HDAC inhibitor effects of rapamycin in vitro also have not provided strong support for acute effects of rapamycin. Short-term exposure of neurons in vitro to rapamycin did not alter neuronal firing under baseline  conditions, and it had limited benefits under conditions of provoked neuronal firing. One way to determine if rapamycin acutely suppresses seizure activity is to compare it to known anticonvulsants. Rapamycin has not been systematically tested in a battery of acute seizure tests like those used routinely to screen candidate therapeutics in preclinical trials. Using similar tests, we found that rapamycin has a limited acute anticonvulsant effect. Furthermore, rapamycin exposure for #6 h has a profile that is comparable to drugs that suppress voltage-gated sodium channel activity. Even when tested for longer times, rapamycin still has an acute seizure test profile that does not match the profiles of either the ketogenic diet or another dietary antiseizure intervention, intermittent fasting. Thus, the anticonvulsant mechanisms of rapamycin may be distinct from other metabolism-based therapies. Because of the adverse effects of rapamycin and related drugs in patients, the design of more specific agents and minimize side effects.
conditions, and it had limited benefits under conditions of provoked neuronal firing. One way to determine if rapamycin acutely suppresses seizure activity is to compare it to known anticonvulsants. Rapamycin has not been systematically tested in a battery of acute seizure tests like those used routinely to screen candidate therapeutics in preclinical trials. Using similar tests, we found that rapamycin has a limited acute anticonvulsant effect. Furthermore, rapamycin exposure for #6 h has a profile that is comparable to drugs that suppress voltage-gated sodium channel activity. Even when tested for longer times, rapamycin still has an acute seizure test profile that does not match the profiles of either the ketogenic diet or another dietary antiseizure intervention, intermittent fasting. Thus, the anticonvulsant mechanisms of rapamycin may be distinct from other metabolism-based therapies. Because of the adverse effects of rapamycin and related drugs in patients, the design of more specific agents and minimize side effects.
There must be a mechanism by which PLP is released to activate the newly synthesized apo-B6 enzymes
We observed that some catalytic activity of PL Perifosine kinase returns upon transfer of the tightly bound PLP to apo-eSHMT but is much less than the expected 50%. Future studies are focused on understanding in much more detail the mechanism of transfer of PLP and the reactivation of PL kinase. There are three known enzymes in living systems that catalyze the production of PLP: PNP oxidase, present in both prokaryotic and eukaryotic organisms; PL kinase, which is also widely distributed in nature; and PLP synthase, which is found in plants and many microorganisms. Both PNP oxidase and PLP synthase have been shown to bind PLP tightly and to transfer the tightly bound PLP to an apo-B6 enzyme. This report is the first study on the properties of the formation and dissociation of a tightly bound PLP in ePL kinase. Classical competitive inhibitors act rapidly and show a high affinity for the active site of the ground state enzyme, whereas slow binding inhibitors show a high affinity for an intermediate state of the enzyme. Slow binding inhibition is characterized by an initial weak binding to the ground state enzyme, followed by tighter binding to the transition state structure. In general, this type of inhibition is considered more physiologically relevant since upstream accumulation of the substrate cannot relieve the inhibition brought about by this form of inhibition. The results presented here suggest that PLP is a slow tight binding inhibitor of ePL kinase. The mechanism of inhibition consists in the formation of a Schiff base between PLP and an active site lysine residue. The inactivation of the enzyme is faster when both PLP and MgADP are present, compared to when PLP is present alone or together with MgATP. Therefore, the inhibition occurs more rapidly during the catalytic turnover of the enzyme, in which the enzyme may go through an intermediate state whose Nutlin-3 conformation favors the covalent binding of PLP. It appears that during the catalytic cycle, or when both PLP and MgADP are bound, the active site of ePL kinase is in a conformation that places the e-amino group of K229 in a favorable position to form a covalent bond with C49 of PLP. The position of K229 in the active site structure of the unliganded ePL kinase is shown in Fig. 6B. Formation of an aldimine between PLP and the e-amino moiety of K229 is suggested by the absorption maximum at 420 nm and by the failure of PLP to bind tightly to K229Q ePL kinase and to inhibit its activity. The 336 nm absorbing band of the tightly bound PLP can be accounted for by several possible structures. One of the most probable is a carbinolamine intermediate, which occurs during the formation of the aldimine. In the carbinolamine structure, the C49 carbon of PLP is tetrahedral because of the addition of the e-amino moiety of K229 across the double bond to oxygen. Another possible structure is the enolimine tautomer of the PLP protonated aldimine also found at the active site of PLP-dependent enzymes. Our results raise questions about the role of ePL kinase in vivo. The observed inhibition mechanism and the transfer of PLP to apo-B6 enzymes may be a strategy to tune ePL kinase activity on the actual requirements of the PLP cofactor. Moreover, since PLP is such a reactive compound, having it bound tightly to ePL kinase would afford protection against unwanted side reactions,in whichit can be dephosphorylated  or form aldimines with free amino acids or eamino groups on lysine residues in non-B6 proteins. We observed that the tightly bound PLP is protected from dephosphorylation by either a specific PLP phosphatase or alkaline phosphatase. But if protecting PLP from the unproductive side reactions is the purpose of its tight binding.
or form aldimines with free amino acids or eamino groups on lysine residues in non-B6 proteins. We observed that the tightly bound PLP is protected from dephosphorylation by either a specific PLP phosphatase or alkaline phosphatase. But if protecting PLP from the unproductive side reactions is the purpose of its tight binding.
Particularly evident in the development of human hepatocellular carcinoma it is highly likely
JNK1 mediates its versatile functions strictly in a cell type-dependent manner. Besides being reported as a PMA-inducible protein, Noxa was originally identified as a p53-induced  stress response gene, but is now known to be regulated by an array of different transcription factors independently of p53. However, although several transcription factors known to participate in JNK signaling and diverse apoptosis pathways, and that were even shown to constitute prominent targets of the proteasome, none of those examined here including c-Jun, c-Myc, Hif1a, ATF3, ATF4 and GR appear to be involved in the herein observed JNK1/2dependent opposite regulation of Noxa. A participation of p53 cannot be completely ruled out, as a few MEF lines studied here may harbor p53 mutations. Perhaps most unexpected was our finding that even the knockdown of c-Myc, a transcription factor that was recently demonstrated to transcriptionally upregulate Noxa during bortezomib-induced apoptosis, did not affect Noxa expression despite the observed protection of JNK22/2 cells from PI-induced apoptosis. Particularly with regard to the fact that JNKs phosphorylate and thereby regulate the apoptotic function of c-Myc, its previous identification as a potent Noxa inducer upon PI treatment provided strong evidence for the existence of a JNK-Myc-Noxa axis, at least in melanoma cells. As the thereby identified c-Myc binding site in the Noxa promoter is conserved among human, mouse and rat, it is presently unknown why c-Myc induces expression of Noxa only in human melanoma cells, but not in the MEF lines studied here. Also cells lacking JNK1 eventually succumb to apoptosis in almost the complete absence of Noxa, albeit in a delayed manner. Together with the observation that knockdown of Noxa had no effect on the delayed JNK1/2-independent cell death pathway occurring most likely in all here examined MEF lines following their exposure to PIs, our results strongly support the existence of alternative PI-induced pathways that kill cells independently of JNKs and Noxa. The BH3-only protein Bim might be part of such a pathway, as it was found by us and others upregulated in response to PIs in a JNK-independent manner, and its knockdown partially protected JNK22/2 cells from PI-induced apoptosis. GSI-IX side effects Furthermore, the PI-mediated upregulation of Bim was, unlike the induction of Noxa, not entirely blocked in the presence of cycloheximide. This suggests that the increase in Bim protein expression is probably a direct effect of INCB18424 proteasome inhibition that prevents degradation of this pro-apoptotic BH3-only molecule. Thus, Bim most likely represents an alternative route to cell death in cases in which PIs are unable to mediate the JNK1-dependent upregulation of Noxa. In summary, we have shown here that a rapid PI-induced apoptosis pathway critically depends on the induction of Noxa that is controlled by JNK1 and JNK2 in an opposing manner. Although we were unable so far to identiy the transcription factor involved, our results might help to further improve future anticancer strategies that are based on proteasomal inhibitors. Thereby, one should keep in mind that our observations are solely based on the use of immortalized MEFs. To exclude possible phenotypical changes acquired during their immortalization, it will be necessary to confirm these findigs using primary MEFs or lymphocytes from JNK1/2 knockout mice.
stress response gene, but is now known to be regulated by an array of different transcription factors independently of p53. However, although several transcription factors known to participate in JNK signaling and diverse apoptosis pathways, and that were even shown to constitute prominent targets of the proteasome, none of those examined here including c-Jun, c-Myc, Hif1a, ATF3, ATF4 and GR appear to be involved in the herein observed JNK1/2dependent opposite regulation of Noxa. A participation of p53 cannot be completely ruled out, as a few MEF lines studied here may harbor p53 mutations. Perhaps most unexpected was our finding that even the knockdown of c-Myc, a transcription factor that was recently demonstrated to transcriptionally upregulate Noxa during bortezomib-induced apoptosis, did not affect Noxa expression despite the observed protection of JNK22/2 cells from PI-induced apoptosis. Particularly with regard to the fact that JNKs phosphorylate and thereby regulate the apoptotic function of c-Myc, its previous identification as a potent Noxa inducer upon PI treatment provided strong evidence for the existence of a JNK-Myc-Noxa axis, at least in melanoma cells. As the thereby identified c-Myc binding site in the Noxa promoter is conserved among human, mouse and rat, it is presently unknown why c-Myc induces expression of Noxa only in human melanoma cells, but not in the MEF lines studied here. Also cells lacking JNK1 eventually succumb to apoptosis in almost the complete absence of Noxa, albeit in a delayed manner. Together with the observation that knockdown of Noxa had no effect on the delayed JNK1/2-independent cell death pathway occurring most likely in all here examined MEF lines following their exposure to PIs, our results strongly support the existence of alternative PI-induced pathways that kill cells independently of JNKs and Noxa. The BH3-only protein Bim might be part of such a pathway, as it was found by us and others upregulated in response to PIs in a JNK-independent manner, and its knockdown partially protected JNK22/2 cells from PI-induced apoptosis. GSI-IX side effects Furthermore, the PI-mediated upregulation of Bim was, unlike the induction of Noxa, not entirely blocked in the presence of cycloheximide. This suggests that the increase in Bim protein expression is probably a direct effect of INCB18424 proteasome inhibition that prevents degradation of this pro-apoptotic BH3-only molecule. Thus, Bim most likely represents an alternative route to cell death in cases in which PIs are unable to mediate the JNK1-dependent upregulation of Noxa. In summary, we have shown here that a rapid PI-induced apoptosis pathway critically depends on the induction of Noxa that is controlled by JNK1 and JNK2 in an opposing manner. Although we were unable so far to identiy the transcription factor involved, our results might help to further improve future anticancer strategies that are based on proteasomal inhibitors. Thereby, one should keep in mind that our observations are solely based on the use of immortalized MEFs. To exclude possible phenotypical changes acquired during their immortalization, it will be necessary to confirm these findigs using primary MEFs or lymphocytes from JNK1/2 knockout mice.