To examine whether VE-821 ATM/ATR inhibitor pitstop 2 is inhibiting CIE through its effects on the Axitinib clathrin N-terminal domain, we looked at transferrin and MHCI endocytosis in cells depleted of clathrin heavy chain or the m2 subunit of the adaptor protein complex AP2, both of which were depleted to approximately 12 and 14% of control levels, respectively. Depletion of the m2 subunit of AP2or of clathrin heavy chainby siRNA results in a block in transferrin endocytosis in most cells while endocytosis of MHCI by CIE is not affected. The addition of pitstop 2 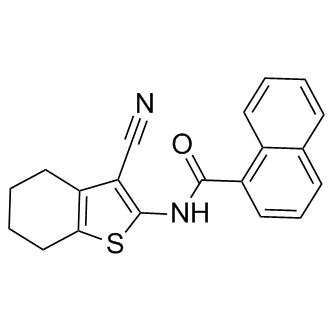 to the m2 and clathrin heavy chain depleted cells still led to a block in endocytosis of MHCI, suggesting that pitstop is blocking CIE through a site independent of clathrin. To gain further insight into how this compound might be blocking CIE, a process that occurs independently of clathrin and dynamin but is sensitive to PM cholesterol levels, we asked whether mobility of cargo proteins entering cells by CIE might be affected by pitstop 2. To do this, we labeled cells expressing SNAPTac with the non-releasable probe, Alexa 488-conjugated BG ligand, and then imaged the cells live before and after photobleaching. In control cells treated with DMSO, surface fluorescence recovered with a t1/2 of approximately 30 sec. In contrast, there was no recovery of fluorescence for the duration of the experiment in cells treated with pitstop 2, suggesting that most of the PM SNAP-Tac was immobile. This dramatic change in surface mobility was also observed for GFP-labeled H-Ras, a marker for the CIE endosomal membrane system. A similar “freezing” of the clathrin and AP2 coat complexes with pitstop 2 was also observed in the original characterization of the compound, suggesting a striking target at the PM that may cause an inhibitory effect for most endocytic events or a general global alteration of PM structure. On the other hand, we did observe that endocytosis of shiga toxin still occurred in cells treated with pitstop 2as was previously reported, although the amount of shiga toxin internalized was less than in controls. Shiga toxin may be more resistant to pitstop as compared to other endogenous CIE cargo proteins due to its ability to bind to and cluster Gb3 glycolipid, forming a tubular invaginated entry structure into cells. Taken together, our findings demonstrate that pitstop 2 cannot be used to determine that a protein enters cells by CDE since it blocks CIE as effectively as CDE. This effect, observed for many endogenous cargo proteins and in all human cell lines examined, is due to a second site of action for the compound since it still inhibits CIE in cells where clathrin has been depleted. This second site of action may explain some of the unusual behavior of cells treated with pitstop as pointed out by Lemmon and Traub. It provides a cautionary tale for the in vivo application of “specific” small molecule inhibitors developed through chemical design as this approach cannot exclude second sites of action in living cells. Improving therapy for patients with Glioblastoma multiformeis one of the biggest challenges in oncology. Although molecular targeting has shown success in many cancers, targeted therapy for GBM has yet to demonstrate an appreciable clinical survival benefit. For example, targeting of Epidermal Growth Factor Receptorwith small molecules or monoclonal antibodies has been reported to offer no survival benefit, despite the fact that EGFR is the most common genomically altered oncogene in GBM, and targeting EGFR has shown benefit in other cancers. So an important question is: can targeted therapy provide a benefit to GBM patients?
to the m2 and clathrin heavy chain depleted cells still led to a block in endocytosis of MHCI, suggesting that pitstop is blocking CIE through a site independent of clathrin. To gain further insight into how this compound might be blocking CIE, a process that occurs independently of clathrin and dynamin but is sensitive to PM cholesterol levels, we asked whether mobility of cargo proteins entering cells by CIE might be affected by pitstop 2. To do this, we labeled cells expressing SNAPTac with the non-releasable probe, Alexa 488-conjugated BG ligand, and then imaged the cells live before and after photobleaching. In control cells treated with DMSO, surface fluorescence recovered with a t1/2 of approximately 30 sec. In contrast, there was no recovery of fluorescence for the duration of the experiment in cells treated with pitstop 2, suggesting that most of the PM SNAP-Tac was immobile. This dramatic change in surface mobility was also observed for GFP-labeled H-Ras, a marker for the CIE endosomal membrane system. A similar “freezing” of the clathrin and AP2 coat complexes with pitstop 2 was also observed in the original characterization of the compound, suggesting a striking target at the PM that may cause an inhibitory effect for most endocytic events or a general global alteration of PM structure. On the other hand, we did observe that endocytosis of shiga toxin still occurred in cells treated with pitstop 2as was previously reported, although the amount of shiga toxin internalized was less than in controls. Shiga toxin may be more resistant to pitstop as compared to other endogenous CIE cargo proteins due to its ability to bind to and cluster Gb3 glycolipid, forming a tubular invaginated entry structure into cells. Taken together, our findings demonstrate that pitstop 2 cannot be used to determine that a protein enters cells by CDE since it blocks CIE as effectively as CDE. This effect, observed for many endogenous cargo proteins and in all human cell lines examined, is due to a second site of action for the compound since it still inhibits CIE in cells where clathrin has been depleted. This second site of action may explain some of the unusual behavior of cells treated with pitstop as pointed out by Lemmon and Traub. It provides a cautionary tale for the in vivo application of “specific” small molecule inhibitors developed through chemical design as this approach cannot exclude second sites of action in living cells. Improving therapy for patients with Glioblastoma multiformeis one of the biggest challenges in oncology. Although molecular targeting has shown success in many cancers, targeted therapy for GBM has yet to demonstrate an appreciable clinical survival benefit. For example, targeting of Epidermal Growth Factor Receptorwith small molecules or monoclonal antibodies has been reported to offer no survival benefit, despite the fact that EGFR is the most common genomically altered oncogene in GBM, and targeting EGFR has shown benefit in other cancers. So an important question is: can targeted therapy provide a benefit to GBM patients?