Its members are conserved from yeast to human. One of the family members, Yap8, appears in Figure 3C and shows accelerated evolution to the average of the genome, which is consistent with the hypothesis that Yap8 is under positive selection to fulfill a wider genetic program required to deal with new environmental stimuli. Reb1 protein is an essential, auto-regulated DNA-binding protein that binds to many sites in the S. cerevisiae genome. Van Slyke et al. showed that Reb1p is likely to play an important role in the complex regulation of CLB2 product 2a protein with a central distinct role among the cyclins2 which will in turn influence the timing or progression of the cell cycle, or the Butenafine hydrochloride budding process. Consequently, identification of other proteins that affect this promoter and their interactions will allow us to understand not only the regulatory circuitry of CLB2, but also the mechanisms by which cell cycle is regulated. Sko1 is also a basic leucine zipper TF of the ATF/CREB family. In a recent breakthrough on our Chlorhexidine hydrochloride understanding on cell cycle control, Niu et al observed that over-expression of the TF Sko1 arrest cells at G1 phase by activating the pheromone response pathway, supporting the notion that many genes may gain function upon over-expression. Surprisingly, the 7 highly interconnected TFs described above, regulate the expression of a gene with unknown function, FMP43, whose expression level was very high under all conditions tested in this study and significantly differentially expressed by FA addition. The identification of FMP43 as one of the primary contributors of the environmental and internal sensing mechanisms to achieve network dynamics attracted our attention. Subsequently, we further explored how the phenotypic biomarkers and the network robustness are affected by redundancy and degeneracy of this protein and its human ortholog. In actively dividing cells, cell size reflects the balance between growth and division. Environmental or genetic perturbations, such as addition of a compound in the culture medium or gene deletions, could shift the balance in favor of growth. Such shift could be either due to higher growth rate of the cells or a delay in division, and it inevitably results in increase of the cell size. These principles apply to the present study, where a gene deletion resulted in higher growth rate, and thus higher cell size and biomass yield for the constructed mutant compared with the wild type. This interpretation also implies that cell cycle progression could be affected. A recent study showed that the ability of yeast cells to grow changes during the cell cycle. Specifically, during the normal cell cycle, cellular growth is slower at the passage through the G1/S-phase boundary, while the ability of cells to grow is higher in anaphaseand G1-arrested cells than in any other cell cycle stage. If cell cycle progression in the FMP mutant is indeed altered, then we hypothesize that FMP43 protein is related to and somehow affects key regulatory mechanisms governing the cell cycle, as for instance the G1/S-phase control point. Additional experiments, to detect any cell cycle dissimilarity of the FMP strain compared to the CON strain, are needed. Flow cytometry experiments 2using the propidium iodide to stain nuclear DNA or a more sensitive DNA stain2 would be suitable to this direction. A plethora of computational approaches can be used to overcome the limitations of experimental techniques. Computational tools have become critical for the integration, representation and visualization of heterogeneous genomics, proteomics and biomedical data. Experimental techniques, like 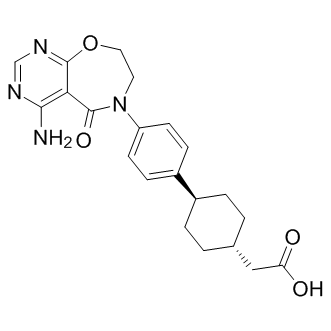 yeast-twohybrid, have enabled to pair-wisely screen protein-protein interactions. Nevertheless, the study of protein complex data involving more than two partners is relatively restricted due to the limitations of the currently available high-throughput techniques. Computational approaches complement experimental methods for the detection of protein complexes using protein interaction data.
yeast-twohybrid, have enabled to pair-wisely screen protein-protein interactions. Nevertheless, the study of protein complex data involving more than two partners is relatively restricted due to the limitations of the currently available high-throughput techniques. Computational approaches complement experimental methods for the detection of protein complexes using protein interaction data.
Month: June 2019
DDB1 involved in NER the nuclear DRP1 level and cisplatin resistance in adenocarcinoma cells
DDB1, which is also involved in NER, is overexpressed in cisplatin resistant cancer cell lines. Elevated glutathione Stransferase P1 expression has been associated with resistance to cisplatin-based chemotherapy in several cancer cell lines. Our gene set comparison analyses demonstrate a significant overlap between the ES cell signatures and our chemotherapy resistance signatures. No prior studies have demonstrated the enrichment of ES cell signatures in clinical samples collected at the time of acquired resistance to cytotoxic chemotherapy. Accumulating evidence suggests an association between a stem cell phenotype and intrinsic chemoresistance. Animal studies have suggested that the cell population exhibiting cancer stem cell characteristics is enriched in xenograft 3,4,5-Trimethoxyphenylacetic acid tumors following chemotherapy. While ES cell signatures may not perfectly reflect the phenotype of gastric cancer stem cells, the enrichment of ES cell signatures in chemoresistant tumors may reflect the survival advantage of tumor cells expressing stem cell regulatory networks. This was validated by our finding that 72 genes shared by the acquired resistance and ES cell signatures were sufficient to predict the initial response to CF. This study has identified a molecular signature for acquired resistance to CF therapy in gastric cancer patients. This signature is able to identify patients likely to have a short or longer term response to CF suggesting it reflects the molecular profile of chemoresistant clones and not non-specific drug effects. Genes contained within this signature, such as Akt/mTOR pathway genes, TRAP1, RAD23A, and GSTP1, may be potentially useful targets for treating tumors resistant to CF therapy. Future studies will be required to confirm these results and to determine whether our novel approach to develop an acquired resistance signature that predicts the therapeutic response of patients to specific chemotherapies is applicable to other types of cancer. A major finding of this study is the identification of a gene signature that emerged in association with tumor resistance to CF therapy in patients who initially benefited from CF therapy. Prior genomic predictors for the chemotherapy response, which were developed using pretreatment tissue samples, have demonstrated a mixed performance. Here we demonstrate that the posttreatment samples collected at the time of acquired resistance, although difficult to obtain clinically, contain unique genomic information that can be used to predict the initial response to cytotoxic chemotherapy. No prior studies have explored acquired resistance using genome-wide analysis of clinical samples, although 2 prior studies evaluated the gene expression pattern in residual disease after the completion of neoadjuvant chemotherapy. Lee, et al. demonstrated that postchemotherapy tumor gene signatures outperforms baseline signatures and clinical predictors in predicting for pathological response and progression-free survival, although these investigators collected posttreatment breast tumors 3 weeks after chemotherapy, not at the time of progressive disease as in our study. Our data is consistent with the aforementioned study that comparing postchemotherapy and prechemotherapy gene expression signatures might be a feasible approach to the identification of predictive signatures. Also, our data provides the first genomic evidence in clinical samples supporting a conventional model for the emergence of acquired resistance whereby resistance emerges through a selective, clonal outgrowth of small 4-(Benzyloxy)phenol populations of pre-existing, chemoresistant tumor cells. While the ”72-gene acquired resistance signature” was developed mainly for potential clinical utility, it contains several overexpressed genes that have been shown to lead to chemoresistance. TRAP1 overexpression leads ![]() to 5-fluorouracil-, oxaliplatin- and irinotecanresistant phenotypes in different neoplastic cells. Silencing of hHR23A, a nucleotide excision repair enzyme, decreases the nuclear DRP1 level and cisplatin resistance in lung adenocarcinoma cells.
to 5-fluorouracil-, oxaliplatin- and irinotecanresistant phenotypes in different neoplastic cells. Silencing of hHR23A, a nucleotide excision repair enzyme, decreases the nuclear DRP1 level and cisplatin resistance in lung adenocarcinoma cells.
Biological roles of the SUMO pathway and also uncover novel connections between sumoylation
Signal transduction, the cell cycle, and development. Furthermore, our SUMO conjugated proteome should serve as a rich resource for those studying the roles of sumoylation in metazoan development. This quantitative review confirmed that RNA and CD4 have very different time patterns of Mechlorethamine hydrochloride clinical prognostic value during untreated HIV-1 infection. Within the first 2 years of infection, RNA immediately gives some indication of long-term prognosis. Due to constant relative risks and constant within-population variability, RNA remains similarly informative when measured during later years. CD4, in contrast, carries little prognostic value over early years. Its within-population variability then instead largely relates to pre-infection CD4 levels, which vary by up to a factor ten among uninfected adults without influencing prognosis after infection. As infection progresses and worsening immune deficiency allows opportunistic infections and AIDS-defining illnesses to occur, the prognostic value of CD4 increases, due to strong increases in relative prognostic risks per unit CD4 decrease and increasing proportional within-population variability in CD4 levels. Part of the prognostic value may reflect that OI themselves temporarily reduce CD4 and increase RNA 2 effects that may be common in clinic populations where the occurrence of OIs is often the reason for diagnosis, especially in Africa. For relative risk studies, data analyzed were Gentamycin Sulfate limited to untreated cohorts and did not address toxicity, viral resistance and cost associated with ART. We nevertheless believe  that the findings are important for clinical decision making, because the prior question that physicians face is their patients�� prognosis if treatment is not initiated. Furthermore, despite our attempts to include only high-quality studies and to focus on standardized outcomes with known covariates, our pooled analyses are not meta-analyses in the strict sense. Notably, included studies varied in rates of loss to follow-up, extent of exposure to antiretroviral mono- or bi-therapy, OI prophylaxis and treatment, and patient inclusion criteria and age ranges; however, available data and statistical power precluded optimal assessment of these possible determinants. The capability of gene expression microarrays to simultaneously measure essentially all human genes has made possible a variety of approaches to analyzing biological samples.
that the findings are important for clinical decision making, because the prior question that physicians face is their patients�� prognosis if treatment is not initiated. Furthermore, despite our attempts to include only high-quality studies and to focus on standardized outcomes with known covariates, our pooled analyses are not meta-analyses in the strict sense. Notably, included studies varied in rates of loss to follow-up, extent of exposure to antiretroviral mono- or bi-therapy, OI prophylaxis and treatment, and patient inclusion criteria and age ranges; however, available data and statistical power precluded optimal assessment of these possible determinants. The capability of gene expression microarrays to simultaneously measure essentially all human genes has made possible a variety of approaches to analyzing biological samples.
We show that N-STZ dams who have the highest incidence of macrosomic newborns also exhibit Folinic
Long-term outcomes for two entire patient cohorts at independent institutions, they should be interpreted with caution as they are derived from retrospective chart reviews. If confirmed, these results make a compelling case for the enhanced use of sensitive diagnostic and predictive tools, including recently described genetic tests, to identify patients most likely to benefit from IFNa-based treatment. Moreover, the potential for adverse outcomes should be considered in current and future studies examining HCV treatment using pegylated-IFNa/RBV in combination with newer agents such as HCV protease inhibitors, as a substantial proportion of null or partial responders with advanced fibrosis will emerge from these treatment groups. In particular, it may be advisable not to retreat these patients with IFNa, but to keep them under observation until IFNa-free regimens are available. Peroxisome proliferator-activated receptors are nuclear receptors that function as ligand-inducible transcription factors. Consistent with their regulation by fatty acids and eicosanoid metabolites, PPARs function as modulators of lipid metabolism and inflammatory responses. The three PPAR subtypes activate their target genes LOUREIRIN-B through binding to PPAR response elements as heterodimers with members of the retinoid receptor family. Genome-wide analyses have identified PPRE-mediated repression as a major mechanism of transcriptional regulation by unliganded PPARb/d, and revealed that a subset of these repressed genes is activated by an agonist-mediated switch.We report that were destined to die from lytic reactivation-induced cell death, but also dysregulated expression of viral and cellular IL6, which have been previously implicated in KSHV tumorigenesis.Further, we noticed that high TUBB3 transcript levels were predictive of poor survival in patients treated with a cumulatively higher dose of ixabepilone. However, since this finding is based on a small sample size it should be interpreted with caution. Whether ixabepilone efficacy is indeed dependent on TUBB3 transcript levels is an interesting question that needs to be answered and validated in larger multi-arm studies. In this study we also investigated MMR status in primary tumors from patients treated with ixabepilone in the metastatic setting, based on previous reports suggesting that taxanes may benefit patients with MMR deficient tumors. This observation appears consistent with clinical studies that show a decrease in the incidence of macrosomia in twin pregnancies complicated with diabetes compared with singleton pregnancies. These data could be indicative of the limitations of the mother in supporting a pregnancy with large fetuses when their number becomes too important. In this case, it could be an adaptive mechanism whose role is decisive for pregnancy success. In the context of pregnancies complicated with diabetes, not only glucose but also maternal lipids may contribute to the risk of having LGA newborns. Clinical studies showed an enhanced insulin resistance in women with gestational diabetes that contributes to significantly increase the lipids concentrations, in particular triglycerides and NEFA, in late gestation. Furthermore, the circulating triglycerides and NEFA concentrations were positively correlated with neonatal weight at birth. There are also studies where no change in plasma lipid levels was found in GDM compared to control pregnant women.
Confirming that all the cells have been sufficiently differentiated and that the two memory B cell populations
We selected probesets to use as the basis of discriminating between cell types by screening for those that offered the most significant differences between the several cells in which they were most highly expressed. In order to optimize the number of Butenafine hydrochloride markers selected, we computed the condition number of matrices of all sizes, from a handful of genes in one extreme, to the whole genome in the other. We observed that the optimal set size was 360 probesets, and we used this set to distinguish between different immune cell subsets and activation states in all subsequent analysis of blood samples. Figure 3 shows some examples of these probesets that discriminate between cell types and are used in deconvolution. Most of these exemplify markers that are relatively specific for one or two cell types. The full collection of basis probesets and their expression levels in all cell types and states are in Table S1. We surveyed the distribution of these data by performing twodimensional hierarchical clustering and Cinoxacin visualized the results as a heatmap with distance-measure dendrograms, and found that the cells all appeared to have distinct expression signatures, to be separated reasonably well on the dendrogram, and to cluster near other samples that we expected to have relatively similar signatures. We examined quantitatively whether the eighteen cell types that we profiled are sufficiently distinct to be resolved by their expression signatures by performing singular value decomposition on the basis matrix and observing the values of the diagonal matrix. This method would yield values at the lower-right corner of the matrix near zero if some of the cells were inadequately different from each other; reassuringly, here the lowest value was 3702.301. Although this value is not considered to be near zero and thus not worrisome, it does represent the aspect of white blood cell biology that we had least successfully resolved, so we explored which cells caused it. We noted that the two memory B cell samples were the two samples that were most similar to each other and we hypothesized that they alone might be responsible for the low end of the SVD diagonal. When we tested this by removing  the IgM memory population from the basis matrix and refactoring it we found that the diagonal very closely resembled the previous diagonal but with the lowest value missing.
the IgM memory population from the basis matrix and refactoring it we found that the diagonal very closely resembled the previous diagonal but with the lowest value missing.