The factors governing the conformational flexibility of the protein remains a key area of interest with regard to the function of this enzyme, and others in the superfamily. In the present study, a coevolution analysis was used to examine sequence relationships of enzymes in the PMM/PGM family. Mutual information analyses were used to identify coevolving residue pairs, and “cliques” were calculated using graph theory to find 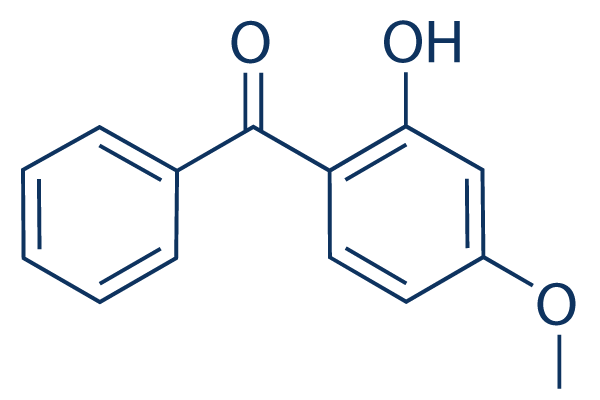 networks of coevolving residues. We identify a tightly linked network of coevolving residues, most of which localize to the interface between domain 4 and the rest of the protein, which is a wellcharacterized site of conformational variability in the enzyme. This result is in distinct contrast to the highly conserved residues in the protein family that cluster in the active site and tend to be directly involved in catalysis/ligand binding. Furthermore, we report the steady-state kinetic characterization of mutants of residues in the coevolving network, and find a reduction in enzyme specificity relative to Atropine sulfate wild-type, ranging from 45 to less than 10%. Mutation of an interface residue that is not part of the coevolving cluster results in no change in specificity, despite making direct structural contacts with other residues in the network selected for mutation. Double mutants of several key residues in the network show additivity in their effects, suggesting that these residues are not significantly coupled energetically. This study sheds new light on the roles of coevolutionary networks in proteins and has implications for the definition of catalytic residues in enzymes, which, as shown here, can be distant from the active site. The ZNMI approach was chosen as it gave results for our MSA that were better when considering both accuracy and reproducibility relative to the other MI methods, using the definitions from. Accuracy of the various methods was assessed by determining if residue pairs with high scores are, on average, close in tertiary structure, the most commonly used metric for evaluating coevolution analysis. ZNMI was calculated between all ungapped positions in the PMM/PGM alignment as described in Methods. The residue couplings from ZNMI were then subjected to a resampling procedure to eliminate errors due to perturbations of the sequence alignment. For this purpose, ensembles of sub-alignments were utilized in a cross validation approach, as described in. For the present study, only residues with 100% reproducibility as determined by the data resampling were considered for further analysis. This stringent cutoff greatly reduces the possibility of false positives from the MI analysis, and, given the large size of the protein, helps reduce the number of coevolving positions to a LOUREIRIN-B manageable number for analysis. It does, however, have the potential disadvantage of missing significant couplings, a limitation that must be considered in interpretation of results. A matrix of the resampled ZNMI scores for all possible residue position pairs in the PMM/PGM MSA is given in Fig. 1A. Also, for comparison, Fig.1B shows per residue plots of the degree, sequence entropy, and gaps for the MSA. The matrix in Fig. 1A is notably sparse due to application of the 100% reproducibility criterion. Despite this, the number of high-scoring residue pairs from the rZNMI analysis is still quite large, and difficult to assess on an individual basis. In general, however, it can be seen that high-scoring couplings are scattered throughout the sequence, although with higher density in the C-terminal half of the protein.
networks of coevolving residues. We identify a tightly linked network of coevolving residues, most of which localize to the interface between domain 4 and the rest of the protein, which is a wellcharacterized site of conformational variability in the enzyme. This result is in distinct contrast to the highly conserved residues in the protein family that cluster in the active site and tend to be directly involved in catalysis/ligand binding. Furthermore, we report the steady-state kinetic characterization of mutants of residues in the coevolving network, and find a reduction in enzyme specificity relative to Atropine sulfate wild-type, ranging from 45 to less than 10%. Mutation of an interface residue that is not part of the coevolving cluster results in no change in specificity, despite making direct structural contacts with other residues in the network selected for mutation. Double mutants of several key residues in the network show additivity in their effects, suggesting that these residues are not significantly coupled energetically. This study sheds new light on the roles of coevolutionary networks in proteins and has implications for the definition of catalytic residues in enzymes, which, as shown here, can be distant from the active site. The ZNMI approach was chosen as it gave results for our MSA that were better when considering both accuracy and reproducibility relative to the other MI methods, using the definitions from. Accuracy of the various methods was assessed by determining if residue pairs with high scores are, on average, close in tertiary structure, the most commonly used metric for evaluating coevolution analysis. ZNMI was calculated between all ungapped positions in the PMM/PGM alignment as described in Methods. The residue couplings from ZNMI were then subjected to a resampling procedure to eliminate errors due to perturbations of the sequence alignment. For this purpose, ensembles of sub-alignments were utilized in a cross validation approach, as described in. For the present study, only residues with 100% reproducibility as determined by the data resampling were considered for further analysis. This stringent cutoff greatly reduces the possibility of false positives from the MI analysis, and, given the large size of the protein, helps reduce the number of coevolving positions to a LOUREIRIN-B manageable number for analysis. It does, however, have the potential disadvantage of missing significant couplings, a limitation that must be considered in interpretation of results. A matrix of the resampled ZNMI scores for all possible residue position pairs in the PMM/PGM MSA is given in Fig. 1A. Also, for comparison, Fig.1B shows per residue plots of the degree, sequence entropy, and gaps for the MSA. The matrix in Fig. 1A is notably sparse due to application of the 100% reproducibility criterion. Despite this, the number of high-scoring residue pairs from the rZNMI analysis is still quite large, and difficult to assess on an individual basis. In general, however, it can be seen that high-scoring couplings are scattered throughout the sequence, although with higher density in the C-terminal half of the protein.