Despite the wide diversity in reproductive strategies, there are characteristic morphological and physiological changes that occur as the oocytes grow and mature. In general, active nuclear transcription and DNA recombination drives meiotic divisions of oogonia during primary growth phases of development. The primary oocyte stage is characterized by the formation of the Mepiroxol follicle including the granulosa cells, which surround the oocyte, the basal lamina, produced by the granulosa layer and the theca cells including blood vessels. Also, one can 
Month: June 2019
Cognitive performance after exposure was associated with a moderate decrease in GFAP staining
An increased mRNA expression level of Grin1, and a trend to an increased generation of nitrites and a negative correlation of those levels to performance of arsenic exposed mice in at least one of the behavioral tasks. Direct in vivo toxic effect of arsenic and metal mixtures on astrocytes with apoptotic cell death and increased ROS generation were demonstrated recently, and an increased Ca2+ release was suggested as an explanation. Since NMDA receptor is directly involved in calcium homeostasis, sustained up-regulation of Grin1, and therefore a possible disruption of the homeostasis, instigated by arsenic exposure during the embryonic life may provide, at least in part, a molecular mechanism for arsenic neurotoxic effects. While the data from our study does not allow correlation of impaired learning in adult mice to changes in expression level and histone modifications, overall the results are urging an association that should be examined in the future with a goal to Folinic acid calcium salt pentahydrate reveal genomewide, and in an unbiased way, changes in expression of genes that may be critical for learning, memory and synaptic plasticity. In recent years arsenic toxicity and its long term effects on human health have been linked to changes in the epigenome. Arsenic is the only in the group of toxic elements that causes changes in all three epigenetic marks �C DNA methylation, histone modifications and expression of noncoding RNAs. Although the results from different studies have not been consistent, changes in DNA methylation are usually linked to arsenic methylation, which is the normal, physiologically induced, pathway for arsenic detoxification in rodents and humans. The sequestration of CH3 groups by arsenic in the detoxification pathway is considered, therefore, one possible molecular mechanism for global and gene-specific DNA hypomethylation, causing changes in the transcriptional activity of genes implicated in the risk for development of certain diseases. It is difficult to reconcile this hypothesis with the results of some in vivo studies, as well as results from epidemiological studies, where DNA hypermethylation has been reported. The methylation pattern and level of methylation on specific lysine residues in the N-terminal histone tails also change in response to arsenic exposure. However, similar to the changes in DNA methylation pattern, a molecular mechanism explaining concurrent increased and decreased methylation of different lysine residues in the animal models, or humans has not been suggested. The first report that arsenic 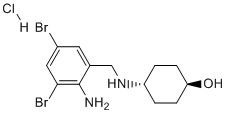 exposure causes a severe reduction in LOUREIRIN-B acetylation of core histones was published in 1983. At this time no clear biological function was assigned to post-translational histone modification. The results with cultured Drosophila cells suggested that the effect of sodium arsenite may be mediated through the activation of heat shock proteins. More recently changes in histone H3 acetylation, and reduced acetylation of H4K16 residue were demonstrated in arsenic treated cultured urothelial and endothelial cells. Elevated histone acetylation responsible for up-regulation of genes involved in apoptosis and stress response has also been reported. The increased global histone acetylation as a result of arsenic exposure in those and other in vitro studies was ascribed to HDAC inhibition. Globally reduced H3K9 acetylation was reported in peripheral mononuclear cells.
exposure causes a severe reduction in LOUREIRIN-B acetylation of core histones was published in 1983. At this time no clear biological function was assigned to post-translational histone modification. The results with cultured Drosophila cells suggested that the effect of sodium arsenite may be mediated through the activation of heat shock proteins. More recently changes in histone H3 acetylation, and reduced acetylation of H4K16 residue were demonstrated in arsenic treated cultured urothelial and endothelial cells. Elevated histone acetylation responsible for up-regulation of genes involved in apoptosis and stress response has also been reported. The increased global histone acetylation as a result of arsenic exposure in those and other in vitro studies was ascribed to HDAC inhibition. Globally reduced H3K9 acetylation was reported in peripheral mononuclear cells.
These results confirmed the specificity of quinacrine for the MglA/SspA heterodimer interface
Currently, Tularemia can be treated with antibiotics such as streptomycin and gentamicin. However, the identification of new therapeutics is very significant, since Francisella can easily be genetically modified and therefore its sensitivity to known antibiotics could be compromised. The manipulation of protein-protein interactions, as targets for therapeutics, is a new and expanding field of research. In this regard, the Francisella MglA/SspA complex is a very attractive system that offers at least three usable interactions: i) with each other, ii) with the RNAP, and iii) with the DNA binding transcription factors FevR and PmrA. Each of these Mepiroxol interactions could potentially be modulated by the action of small molecules. In fact, it was reported that the levels of ppGpp Lomitapide Mesylate modulate the activity of PigR and its interactions with MglA/SspA/RNAP complex in vivo. However, no evidence is available to confirm that the effect is due to a direct interaction with the FevR regulator, the MglA/SspA heterodimer, the RNAP, or due to altered levels of an unknown intracellular metabolite. Here we report the identification of a small molecule that specifically modified MglA and SspA interactions in vitro and in vivo. Using structure guided site directed mutagenesis, we were able to determine that quinacrine hydrochloride binds in the “cleft region” formed by the MglA/SspA heterodimer. The biochemical evidence provided herein suggests that a biologically relevant molecule may act to modulate the expression of pathogenicity determinants in F. novicida, and provide the putative binding residues for such interactions. The identification of small molecules that modulate proteinprotein interactions is an expanding field with important therapeutic applications. We used a fluorescence-based thermal screening assay to uncover chemicals that bind the MglA/SspA heterodimer interface. Though small by today’s standards, the Prestwick chemical library was used to identify several compounds that bind Ft-MglA and Ec-SspA. Of the 1152 compounds present in the Prestwick library, 13 small molecules stabilized Ft-MglA in vitro. The number of positive hits obtained is similar to other transcription factors previously screened in our laboratory. A secondary confirmation assay was designed to link the thermal screening results to the biological function of the target protein. Since MglA and SspA form a heterodimer to bind the RNA polymerase, we used a bacterial two hybrid system as a secondary assay. The rationale was to determine which of the 13 small molecules modulate MglA and SspA interactions. Interestingly, we found that only one compound, quinacrine, decreased the  interaction of Ft-MglA and Ft-SspA by more than 50%. The other 12 compounds examined in the two hybrid system were determined to be biologically insignificant, as no major reduction in MglA/SspA interaction was observed. The thermal stabilization effect observed with these compounds was most likely due to unspecific binding at locations other than the heterodimer interface. Another possibility, however, is a deficiency in the uptake of those chemicals by the E. coli reporter strain. The decrease in MglA/SspA interactions observed in the two hybrid system, in the presence of quinacrine, was correlated with impaired intramacrophage survival of F. novicida and decreased expression of the FPI genes. An interesting observation was that F. novicida could tolerate rather high concentrations of quinacrine when cultured in liquid media, while 25 uM was sufficient to decrease F. novicida macrophage infection and intramacrophage survival.
interaction of Ft-MglA and Ft-SspA by more than 50%. The other 12 compounds examined in the two hybrid system were determined to be biologically insignificant, as no major reduction in MglA/SspA interaction was observed. The thermal stabilization effect observed with these compounds was most likely due to unspecific binding at locations other than the heterodimer interface. Another possibility, however, is a deficiency in the uptake of those chemicals by the E. coli reporter strain. The decrease in MglA/SspA interactions observed in the two hybrid system, in the presence of quinacrine, was correlated with impaired intramacrophage survival of F. novicida and decreased expression of the FPI genes. An interesting observation was that F. novicida could tolerate rather high concentrations of quinacrine when cultured in liquid media, while 25 uM was sufficient to decrease F. novicida macrophage infection and intramacrophage survival.
Providing an excellent model system for agricultural and biological research
Despite these efforts, the precise annotation of these alternatively spliced MUC2 transcripts remains incomplete, and the length of the PTS domain, which is predicted to span 55�C110 cassettes, remains highly polymorphic within the human population. Although the biological relevance of these alternatively spliced products in human is not fully understood, it is believed that they are associated with pathogenesis of intestinal diseases. Although functional studies in mice have indicated that Muc2 plays roles in the biology and health of the gut, the function of the PTS domain in mice is less clear, due to the annotation of a relatively short and imprecise repetitive cassette. Evolutionary studies predict that the gel forming mucins share a common ancestor with lower metazoa, as their domain structures are well conserved across a wide range of species from invertebrates to humans. However, relatively few MUC genes have been identified in avians and amphibians. The first Mucin gene cloned in chicken was ovomucin alpha-subunit, now annotated as MUC5B. In silico predictions and annotation of short mRNAs and expressed sequence tags have generated a putative partial MUC2 cDNA in chicken. However, these studies have provided very little functional annotation evidence of the 3,4,5-Trimethoxyphenylacetic acid genomic organization of the chicken MUC2 locus. To determine the structure, expression, biosynthesis and gene signatures of intestinal mucins from a functional and evolutionary perspective, we cloned the chicken MUC2 cDNA that encodes the MUC2 peptide backbone. We achieved this by analyzing and assembling more than 85 cDNA clones that were generated by overlapping RT-PCR products, rapid amplification of cDNA ends, sequencing of ESTs, and incorporating functional annotation data from the UCSC database and NCBI. We also Folinic acid calcium salt pentahydrate compared our sequence to the predicted chicken cDNA. We found that the 11,359 bp chicken cDNA spans 74.5 kb of genomic DNA and is comprised of at least 64 exons. MUC2 is expressed in multiple regions of the gastrointestinal tract, and we detected transcripts as early as embryonic day 14.5. We found several alternatively spliced products, and characterized the splice junctions of one of these transcripts. We determined that the chicken MUC2 protein is remarkably similar to human and mouse outside of the central PTS domain, but is  highly divergent within this central repetitive structure. In humans, this PTS domain is highly glycosylated by Oglycans in the Golgi, and it is predicted that these posttranslational modifications largely contribute to the innate immune response, as proteolytic cleavage of these sugar chains occurs in the outer mucus layer when these molecules come into contact with foreign pathogens. It will be interesting to compare the posttranslational modifications in chicken with other species, especially given the high degree of divergence in this region. It has been over two decades since the initial cloning of the first intestinal mucin gene in humans. Although the physiological implications and disease associations of mucins on various mucosal surfaces have been well recognized, many questions remain as to how and why the gene architecture of this family contributes to diverse protein modifications that may display distinct functionalities. Different species demonstrate structural and sequence conservations as well as their own uniqueness. Chicken, the moststudied and characterized avian species, bridges the evolutionary gap between mammals and non-amniote vertebrates.
highly divergent within this central repetitive structure. In humans, this PTS domain is highly glycosylated by Oglycans in the Golgi, and it is predicted that these posttranslational modifications largely contribute to the innate immune response, as proteolytic cleavage of these sugar chains occurs in the outer mucus layer when these molecules come into contact with foreign pathogens. It will be interesting to compare the posttranslational modifications in chicken with other species, especially given the high degree of divergence in this region. It has been over two decades since the initial cloning of the first intestinal mucin gene in humans. Although the physiological implications and disease associations of mucins on various mucosal surfaces have been well recognized, many questions remain as to how and why the gene architecture of this family contributes to diverse protein modifications that may display distinct functionalities. Different species demonstrate structural and sequence conservations as well as their own uniqueness. Chicken, the moststudied and characterized avian species, bridges the evolutionary gap between mammals and non-amniote vertebrates.
This hypothesis was comparing the gene expression of the identified specific genes in term placentas of normal and complicated
Genes regulating cell cycle, differentiation and motility, macromolecule biosynthetic and metabolic process, and angiogenesis are up-regulated in early pregnancy, whereas genes involved in lipid and chemosensory metabolism, stress response, signal transduction and ion transport are highly expressed in term placenta. Another critical time-point in human pregnancy and placental function is the switch from early to mid-gestation. In early pregnancy, normal trophoblast development is the key for successful implantation and formation of maternal-fetal interface that facilitates the dialogue between the two organisms. Midgestation placenta supports proportional fetal growth, organ development and fine-scale differentiation, as well as continuing maternal adaptation to pregnancy. Only one published study has compared gene expression differences between first and second trimester placentae and reported 61 differentially expressed loci that have been implicated in pregnancy, reproductive physiolology and interaction between organisms. However, the authors were focused on microarray data analysis, which was not followed by any experimental confirmation or biomedical implication. The present study had three main aims: transcriptome profiling of genes with significant expressional changes in placenta in progression from early to mid-pregnancy, and experimental confirmation of mid-gestation specific gene expression using Taqman qPCR in an extended sample; investigation of the novel hypothesis that the normal course of late pregnancy may be affected when the genes characteristic to mid-gestation placental transcriptome remain highly expressed until term; exploring the protein expression of the most prominent identified genes in pregnancy complications, and pilot evaluation of their applicability as biomarkers. The study reports 154 placental transcripts with significant change in expression levels from gestational weeks 5 to 18. The major advancement in the present study in contrast to earlier publications is the experimental validation of 24 loci from microarray analysis. Furthermore, we confirmed highly significant distinct mid-gestational peaks of gene expression for 10 genes in an extended sample-set of first, second and term placentae. In support of our study hypothesis, several mid-gestation genes Albaspidin-AA exhibited aberrant placental expression in complicated pregnancies at term, and alterations at the protein level were confirmed for CCNG2, LYPD6 and STC1. Our findings have direct potential for biomedical implications as we demonstrate highly significant elevated concentration of circulating hormone STC1 in the maternal serum in PE patients and LOUREIRIN-B especially in the PE subgroup accompanied by affected fetal growth. This warrants further investigations of STC1 as a prognostic biomarker of pregnancy outcome. As an additional novel finding, most of the identified genes specifically up-regulated in mid-gestation placenta have also been implicated in adult complex diseases. We hypothesized that the normal course of late pregnancy may be affected when the genes characteristic to mid-gestation placental transcriptome remain highly expressed until term. The hypothesis is based on the assumption that late pregnancy complications may be accompanied by either of the two scenarios: insufficient down-regulation of mid-gestation specific genes that would normally undergo rapid 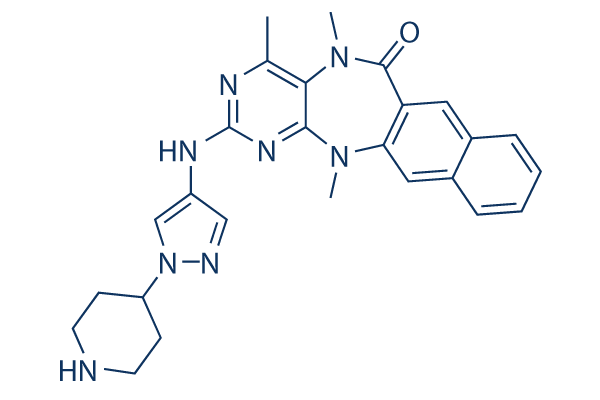 inhibition at term, or continued transcriptional up-regulation of genes that would normally reach their expressional plateau already in mid-pregnancy.
inhibition at term, or continued transcriptional up-regulation of genes that would normally reach their expressional plateau already in mid-pregnancy.