Our drug development scheme should be applicable to the design of small molecules capable of specifically interfering with many other well-characterized inter or intra-molecular interactions with amenable surfaces. Other non-peptide small molecules that LY2109761 disrupt specific protein-protein interactions have been successfully developed in recent times, and they are beginning to show great promise for the therapy of human cancer. In practical terms, we have developed small molecules, and we have shown that these compounds can be used in combination therapy schemes as antitumoral agents in cultured and animal models of cancer. Specifically, compound Ia sensitizes PC-3 prostate cancer cells to the antiproliferative activity of doxorubicin in cultured cells and it shows direct antitumoral activity in mouse tumor xenografts. A number of mechanisms can be at play to cause increased sensitivities of tumor cells to chemotherapy or radiotherapy, including inhibition of NF-kB, downregulation of transporters of the MDR family or the Akt-mTOR pathway. The evidence provided here suggests that at least two mechanisms may be relevant for the increased sensitivity to doxorubicin caused by compound Ia, namely inhibition of NFk-B activity and compromise of DNA repair. The demonstration that this compound disrupts the interaction between Uev1 and Ubc13 provides a mechanistic explanation for its inhibitory activity on the NF-kB signaling pathway. Recently, it has been shown that another ubiquitin conjugating enzyme, UbcH5, can promote K63 polyubiquitylation, and that NF-kB activation by IL-1b is much more strongly dependent on Ubc13-dependent K63 polyubiquitylation than activation by TNF-a. However, a large body of literature strongly suggests a critical role of Ubc13 and K63 polyubiquitylation in the activation of NF-kB not only by IL-1b but also by TNF-a. In this regard, the chain type of ligand-induced ubiquitylation by cIAP of TNF-R1 complex components has not been determined, and, given the recruitment of Ubc13 by cIAP, it is quite possible that 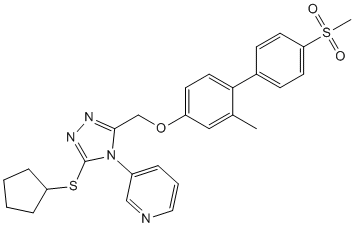 such chains are of the K63 type. Furthermore, mice haploinsuficient for Ubc13 display cell-typespecific defects in chemokine and NF-kB signaling, supporting a critical role of Ubc13 and K63 polyubiquitylation in the activation of NF-kB by different stimuli in vivo, including TNF-a and LPS. Our observations showing that the small molecule antagonist of Ubc13-Uev interactions compound Ia inhibits NF-kB activation by TNF-a would also support a role for Ubc13 in this pathway. Alternative explanations would include the possibility that our compounds inhibit other ubiquitin conjugating enzymes or additional components of the TNF-a signaling BMN673 cascade, which has not been formally ruled out in the present study. On the other hand, it has also been shown that unanchored K63-linked polyubiquitin chains are essential for the activation of the RIG-I pathway in response to viral infection, and that both Ubc13 and Ubc5 are required in this pathway. Therefore, the inhibition of Ubc13 by small compounds could limit the response to viral infections mediated through this pathway. Regarding the role of Ubc13 and K63 polyubiquitylation in DNA damage response, the very high similarity of Uev2 to Uev1, and the computed interaction of compound Ia on the hydrophobic pocket of Ubc13, allows to predict with sufficient confidence that this compound should disrupt also the interaction of Uev2 with Ubc13.
such chains are of the K63 type. Furthermore, mice haploinsuficient for Ubc13 display cell-typespecific defects in chemokine and NF-kB signaling, supporting a critical role of Ubc13 and K63 polyubiquitylation in the activation of NF-kB by different stimuli in vivo, including TNF-a and LPS. Our observations showing that the small molecule antagonist of Ubc13-Uev interactions compound Ia inhibits NF-kB activation by TNF-a would also support a role for Ubc13 in this pathway. Alternative explanations would include the possibility that our compounds inhibit other ubiquitin conjugating enzymes or additional components of the TNF-a signaling BMN673 cascade, which has not been formally ruled out in the present study. On the other hand, it has also been shown that unanchored K63-linked polyubiquitin chains are essential for the activation of the RIG-I pathway in response to viral infection, and that both Ubc13 and Ubc5 are required in this pathway. Therefore, the inhibition of Ubc13 by small compounds could limit the response to viral infections mediated through this pathway. Regarding the role of Ubc13 and K63 polyubiquitylation in DNA damage response, the very high similarity of Uev2 to Uev1, and the computed interaction of compound Ia on the hydrophobic pocket of Ubc13, allows to predict with sufficient confidence that this compound should disrupt also the interaction of Uev2 with Ubc13.