However, as outlined in the Introduction, other investigators have found the opposite effect in gal-3 null mice with NASH in aging mice or induced by the CDAA diet. While there is no obvious explanation for the different findings of these two groups, the preponderance of data from other gal-3 null mouse studies on fibrosis in liver, kidney, lung, and heartdemonstrates that gal-3 appears to be integral to accumulation of fibrosis in INCB18424 parenchymal tissue. In addition, while 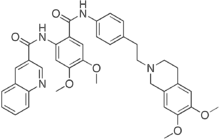 the use of gal-3 null mice is a powerful biological model, there are some drawbacks including the obvious potential impact of background mouse strain. Also, both intracellular and extracellular gal-3 is eliminated in the null mice. The extracellular effects of galectins are related to their lectin properties to bind to glycoproteins whereas their intracellular effects are related to protein-protein interactions. Treatment with GR-MD-02, a complex carbohydrate with galactose residues would be expected to interfere with lectin effects predominantly on the cell surface and in the extracellular space. While the degree of collagen deposition in the model of NASH used in these studies is modest, as in many animal models of NASH, we have previously reported evidence that the same drug agents are effective in reducing much greater degrees of fibrosis and cirrhosis in thioacetamide-treated rats. In these previous studies, the regression in cirrhosis was associated with a reduction in portal hypertension, demonstrating that the change in liver architecture has a physiological effect on liver blood flow and/or resistance. Therefore, it appears that these drugs have effects on the early pathophysiological collagen deposition in NASH, ncluding peri-central and peri-sinusoidal deposition of collagen and potentially later stages of fibrosis and cirrhosis. The presumed proximate mechanism of action of the drugs used in this study is related to gal-3 binding. Gal-3 has a carbohydrate recognition domainwhich is shared among galectin proteins, but in contrast to other galectin proteins, it has a long N-terminal domain that is involved in forming multimers. Gal-3 binds poorly to single galactose molecules, more avidly to galactose containing disaccharides, and most avidly to larger molecules such as glycoproteins with galactose residues. We have shown that our carbohydrate drugs bind to the gal-3 CRD through somewhat different sets of amino acid residues and the affinity at 50% saturation of GR-MD02 and GM-CT-01 to gal-3 is 2.9 mM and 2.8 mM, respectively. This compares to gal-1 binding affinities for GR-MD-02 and GM-CT-01 of 8 mM and 10 mM, respectively. Although galectins are defined by their ability to bind to model carbohydrates containing galactose, such as N-acetyllactosamine, individual galectins appear to bind to different sets of glycans on glycoproteins, thus providing specificity between galectins. For example, galectin-1 and galectin-3 bind to distinct cell surface receptors on T-cells. There are many reported potential ligands for the lectin properties of galectin-3 including laminin, U0126 integrins, collagens, fibronectin, elastin, mucins, CD4+, CD8+, TGFBR, and many others. Binding of galectin-3 to Nglycans has been connected to multiple cellular processes including cell adhesion and migration, immune cell function, inflammation, and neoplasia. It is likely, that inhibition of galectin-3 modulates multiple protein interactions in the extracellular space thereby altering cellular function. We have not determined in these studies which gal-3 protein interactions are abrogated by drug treatment. However, we have data that suggest some downstream processes that are affected, one of which seems to involve macrophages.
the use of gal-3 null mice is a powerful biological model, there are some drawbacks including the obvious potential impact of background mouse strain. Also, both intracellular and extracellular gal-3 is eliminated in the null mice. The extracellular effects of galectins are related to their lectin properties to bind to glycoproteins whereas their intracellular effects are related to protein-protein interactions. Treatment with GR-MD-02, a complex carbohydrate with galactose residues would be expected to interfere with lectin effects predominantly on the cell surface and in the extracellular space. While the degree of collagen deposition in the model of NASH used in these studies is modest, as in many animal models of NASH, we have previously reported evidence that the same drug agents are effective in reducing much greater degrees of fibrosis and cirrhosis in thioacetamide-treated rats. In these previous studies, the regression in cirrhosis was associated with a reduction in portal hypertension, demonstrating that the change in liver architecture has a physiological effect on liver blood flow and/or resistance. Therefore, it appears that these drugs have effects on the early pathophysiological collagen deposition in NASH, ncluding peri-central and peri-sinusoidal deposition of collagen and potentially later stages of fibrosis and cirrhosis. The presumed proximate mechanism of action of the drugs used in this study is related to gal-3 binding. Gal-3 has a carbohydrate recognition domainwhich is shared among galectin proteins, but in contrast to other galectin proteins, it has a long N-terminal domain that is involved in forming multimers. Gal-3 binds poorly to single galactose molecules, more avidly to galactose containing disaccharides, and most avidly to larger molecules such as glycoproteins with galactose residues. We have shown that our carbohydrate drugs bind to the gal-3 CRD through somewhat different sets of amino acid residues and the affinity at 50% saturation of GR-MD02 and GM-CT-01 to gal-3 is 2.9 mM and 2.8 mM, respectively. This compares to gal-1 binding affinities for GR-MD-02 and GM-CT-01 of 8 mM and 10 mM, respectively. Although galectins are defined by their ability to bind to model carbohydrates containing galactose, such as N-acetyllactosamine, individual galectins appear to bind to different sets of glycans on glycoproteins, thus providing specificity between galectins. For example, galectin-1 and galectin-3 bind to distinct cell surface receptors on T-cells. There are many reported potential ligands for the lectin properties of galectin-3 including laminin, U0126 integrins, collagens, fibronectin, elastin, mucins, CD4+, CD8+, TGFBR, and many others. Binding of galectin-3 to Nglycans has been connected to multiple cellular processes including cell adhesion and migration, immune cell function, inflammation, and neoplasia. It is likely, that inhibition of galectin-3 modulates multiple protein interactions in the extracellular space thereby altering cellular function. We have not determined in these studies which gal-3 protein interactions are abrogated by drug treatment. However, we have data that suggest some downstream processes that are affected, one of which seems to involve macrophages.