The resulting active enzyme is a dimer, wherein each subunit contains a p10 and p20 chain and one active site. The caspase enzymatic mechanism is similar to other cysteine proteases; substrate binds to the active site to form the Michaelis complex, a covalent tetrahedral intermediate is formed by attack of the active-site thiolate cysteine on the scissile carbonyl, the substrate amide bond is cleaved to generate an acyl enzyme intermediate, and the intermediate is hydrolyzed by water to yield the new substrate C-terminus and apo-enzyme. Active caspases are capable of cleaving numerous cellular proteinsand carrying out the terminal phase of cell death signaling. Due to the role of caspase-6 in neurodegeneration, there is strong interest in developing selective, small-molecule inhibitors of this enzyme. This family of proteases has proven resistant to traditional methods of drug discovery, however, and most known inhibitors contain a covalent warhead, significant peptidic character, and/or an aspartic acid. Each of these characteristics reduces the potential for caspase selectivity, cell permeability, and blood-brain barrier penetrance. For instance, the traditional caspase probes used in biological assays are tetrapeptides containing the ideal substrate sequences for each caspase and a covalent warhead that reversibly or irreversibly modifies the active-site cysteine. These tools lack the necessary caspase selectivity profiles to facilitate the delineation of isoformspecific signaling pathways in a cellular context. To address these challenges, a number of alternative chemical approaches have been used. Leyva, et al, recently disclosed the design of novel, BMS-907351 nonpeptidic inhibitors identified through “substrate assisted screening”; while potent, these compounds are non-selective and still contain an irreversible covalent warhead. There has also been significant interest in developing noncompetitive or allosteric inhibitors, with the idea that non-active site binding could achieve greater selectivity and improved physicochemical properties over competitive inhibitors. This notion is supported by the discovery of an allosteric site at the dimer interface of caspases 1, 3, and 7. Applying the disulfide-trappingmethod of fragment discovery, scientists at Sunesis Pharmaceuticals identified fragments that bound at the dimer interface and inhibited enzymatic activity. These fragments were not tested for cellular activity, and the druggability of this site remains an interesting, open question. Using a fluorogenic assay platform we identified a series of molecules that inhibit caspase-6 in an unexpected and mechanistically uncompetitive fashion. Detailed structural and mechanistic studies with the most potent of these compounds 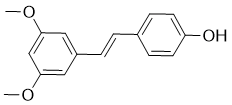 indicate that it binds to the enzyme-substrate complex in a highly specific manner to inhibit substrate turnover. This uncompetitive mechanism of enzyme inhibition is novel for any of the caspase family members. The present compound demonstrates a very distinctive molecular recognition for caspase-6/VEID peptides, and points the way towards utilizing uncompetitive inhibition as a strategy for the discovery of highly selective caspase inhibitors. Our search for caspase-6 inhibitors led to the identification of a highly selective molecule that Compound Library inhibits the enzyme via a novel mechanism not previously described for any of the caspases. Although it has recently been demonstrated for another cysteine protease that the acyl-enzyme intermediate is the primary resting state during the catalytic cycle, stabilization of this intermediate by 3 can be ruled out as the sole mechanism of inhibition, since no fluorophore dependence would be expected if this were the case.
indicate that it binds to the enzyme-substrate complex in a highly specific manner to inhibit substrate turnover. This uncompetitive mechanism of enzyme inhibition is novel for any of the caspase family members. The present compound demonstrates a very distinctive molecular recognition for caspase-6/VEID peptides, and points the way towards utilizing uncompetitive inhibition as a strategy for the discovery of highly selective caspase inhibitors. Our search for caspase-6 inhibitors led to the identification of a highly selective molecule that Compound Library inhibits the enzyme via a novel mechanism not previously described for any of the caspases. Although it has recently been demonstrated for another cysteine protease that the acyl-enzyme intermediate is the primary resting state during the catalytic cycle, stabilization of this intermediate by 3 can be ruled out as the sole mechanism of inhibition, since no fluorophore dependence would be expected if this were the case.